{kind=link}
{kind=link}
{kind=link}
The goal of this python package is to accurately align and quantify viral or bacterial (pathogen) derived reads for 10x single cell data. This software integrates pathogen UMI counts and pathogen UMI gene counts into 10x files generated by first running cellranger count which aligns reads to your host genome of interest. This generates output files for each sample such as features.tsv.gz and matrix.mtx.gz in the filtered_feature_bc_matrix and raw_feature_bc_matrix folders. scPathoQuant then takes the unaligned reads from the host genome and maps them to your pathogen genome of interest and then integrates them back into the cellranger files. This allows softwares such as seurat to be used for easy analysis of the data. The software uses outside software samtools, bowtie2, and htseq to quantify pathogen reads. Default parameters are used when aligning reads to the pathogen genome and and non default htseq-count include using the --intersection-nonempty parameter.
git clone https://github.com/galelab/scPathoQuant.git
cd scPathoQuantthen either
conda env create -f condaenv.yml -n scpathoquant
conda activate scpathoquant
pip install --no-deps .or build a Docker image locally:
docker build -t scpathoquant .Set the following parameters
- -10x = Path/to/10x/sample/ (this the path to the output folder generated by first running
cellranger count) - -op = path/for/results
- -p = number of processors (defualt = 1)
- -p2genome = path/to/pathogenreferencegenome/ - in this folder should be at most 2 files 1) the fasta file with the pathogen reference genome sequence and 2) pathogen gtf file (not mandatory this can just run with fasta file with the pathogen genome). Note: In the fasta file the header will be used to quantify the number of pathogen total reads, it is recommended that if the fasta header is a complicated name it be simplified (i.e. > HIV_virus). The Sars-CoV-2 reference genome and gtf file can be found here
- -align = alignment tool bbmap (default) or bowtie2
- --bbmap_params = parameters specific to the bbmap alignment tool
- --bowtie2_params = parameters specific to the bowtie2 alignment tool
- --tmp_removal = if specified will remove the temporary directory (_tmp/) of files used by scpathoquant (these files can be large so if space is an issue these should be deleted)
Example runs (without Docker):
scpathoquant -10x path/to/10x/sample -op path/for/results -p 8 -p2genome path/to/pathogen/fastafilefolderscpathoquant -10x path/to/10x/sample -op path/for/results -p 8 -p2genome path/to/pathogen/fastafilefolder --tmp_removalscpathoquant -10x path/to/10x/sample -op path/for/results -p 8 -p2genome path/to/pathogen/fastafilefolder --bbmap_params="--semiperfectmode"scpathoquant -10x path/to/10x/sample -op path/for/results -p 8 -p2genome path/to/viral/fastafilefolder -align bowtie2 --bowtie2_params="--very-sensitive --non-deterministic"Example runs (with Docker):
docker run --rm -v path/to/10x/sample:/app/input -v path/to/pathogen/fastafilefolder:/app/genome -v path/for/results:/app/output scpathoquant -10x /app/input -op /app/output -p 8 -p2genome /app/genomedocker run --rm -v path/to/10x/sample:/app/input -v path/to/pathogen/fastafilefolder:/app/genome -v path/for/results:/app/output scpathoquant -10x /app/input -op /app/output -p 8 -p2genome /app/genome --tmp_removaldocker run --rm -v path/to/10x/sample:/app/input -v path/to/pathogen/fastafilefolder:/app/genome -v path/for/results:/app/output scpathoquant -10x /app/input -op /app/output -p 8 -p2genome /app/genome --bbmap_params="--semiperfectmode"docker run --rm -v path/to/10x/sample:/app/input -v path/to/viral/fastafilefolder:/app/genome -v path/for/results:/app/output scpathoquant -10x /app/input -op /app/output -p 8 -p2genome /app/genome -align bowtie2 --bowtie2_params="--very-sensitive --non-deterministic"Output files by scPathoQuant
- pathogen_copy.png - violin plot showing number of cells with pathogen and UMI counts
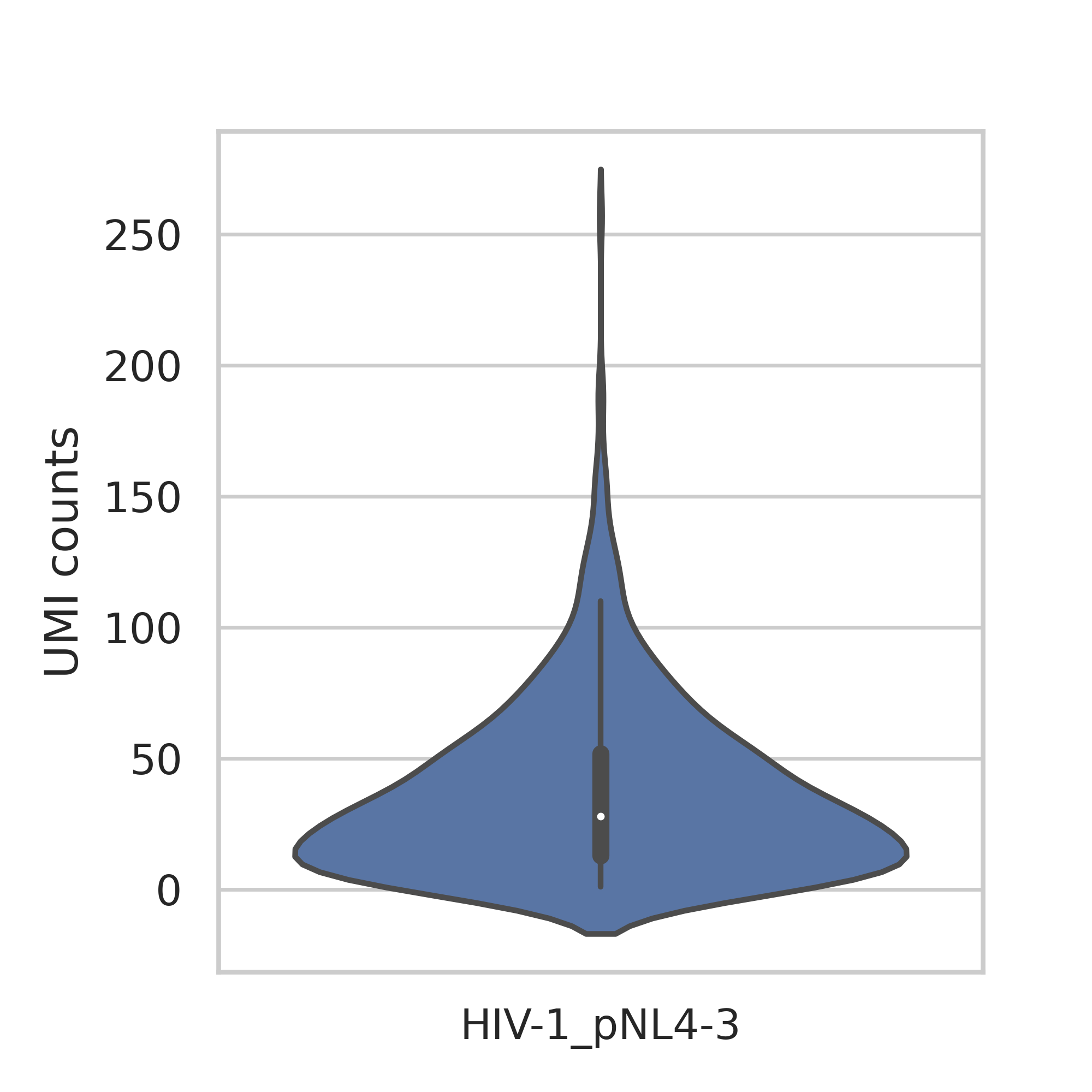
- pathogen_genes.png - violin plot showing cells with pathogen gene UMI counts

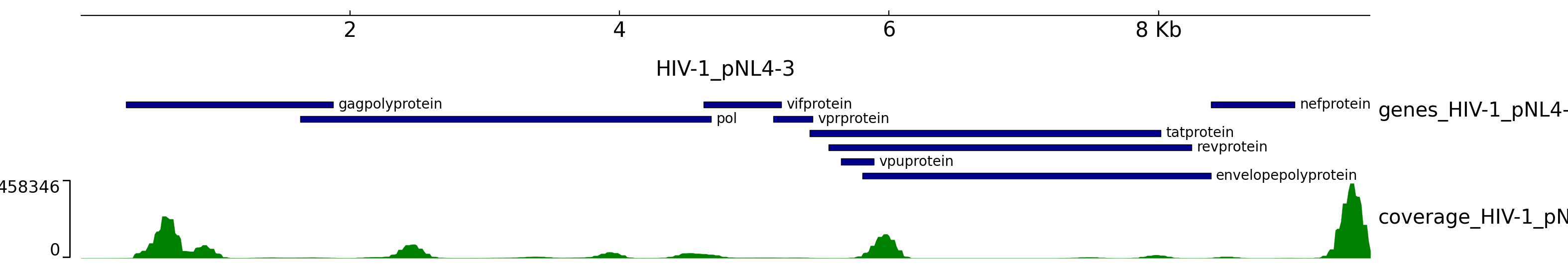
- coveragemap.png - violin plot showing cells with pathogen gene UMI counts
- pathogen_al_counts.csv - total number of reads mapping to the pathogen in each cell
- pathogen_al_gene_counts.csv - number of reads mapping to pathoen genes in each cell
- pathogen_al.bam - reads mapped to pathogen
- pathogen_al_mapped.bam - reads mapped to only pathogen (no unmapped reads)
- pathogen_al_mapped_sort.bam - sorted reads mapped to pathogen (can be used to visualize reads in IGV)
- pathogen_al_mapped_sort.bam.bai - index file to pathogen_al_sort.bam
- pathogen_al_mapped.fq.gz - fastq file of mapped reads to pathogens of interest. Can be used to further perform phylogenetic and clade analyses
- pathogen_al_sort_counts.sam - htseq output reads mapping to pathogen
- pathogen_genes_al_sort_counts.sam - htseq output reads mapping to individual pathogen genes (will not be produced if pathogen/viral gtf is not provided)
- Overwrites original 10x data provided to include pathogen counts and pathogen gene counts (if gtf file is provided)
- filtered_feature_bc_matrix - files that can be integrated into seurat with pathogen counts
- raw_feature_bc_matrix - files that can be integrated into seurat with pathogen counts (this is not always needed)
Example seurat command in R
seurat.object.data <- Read10X(data.dir ="output_scPathoQuant/filtered_feature_bc_matrix")Files in _tmp/ folder
- unmmaped.bam - all unmapped reads from CellRanger (pulled from possorted_genome_bam.bam)
- unmmaped.sam - sam file generated from unmmaped.bam so that barcodes and umis can be extracted for unmmaped reds
- barcodes_umi_read_table.csv - table of unmapped reads and corresponding barcodes and UMIs
- unmapped.fq.gz - all unmapped reads in fastq format could be used fo downstream phylogentic analysis
Examples and test data sets and codes for scPathoQuant can be found here
Whitmore LS, Tisoncik-Go J, Gale M Jr. scPathoQuant: a tool for efficient alignment and quantification of pathogen sequence reads from 10× single cell sequencing datasets. Bioinformatics. 2024 Mar 29;40(4):btae145. doi: 10.1093/bioinformatics/btae145. PMID: 38478395; PMCID: PMC10990681.
BSD - 3-Clause Copyright 2023 University of Washington