![]()




This repository contains the R package now hosted on
Bioconductor
and our stable and development GitHub versions.
(Mac OS Users Only:) Ensure you have installed XQuartz first.
Make sure you have the latest R version and the latest BiocManager
package installed following these
instructions (if you use legacy
R versions (<=3.5.0) refer to the instructions at the end of the
mentioned page).
## install BiocManager if not installed
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
## ensure the following returns TRUE, or follow guidelines
BiocManager::valid()You can then install mixOmics using the following code:
## install mixOmics
BiocManager::install('mixOmics')Install the latest stable version (see below for latest
development
version) of mixOmics from GitHub (as bug-free as it can be):
BiocManager::install("mixOmicsTeam/mixOmics") Check after installation that the following code does not throw any error (especially Mac users - refer to installation instructions) and that the welcome message confirms you have installed the latest version:
library(mixOmics)
#> Loaded mixOmics ?.?.?You can also install the development version for new features yet to be widely tested (see What’s New):
BiocManager::install("mixOmicsTeam/mixOmics@devel")We welcome community contributions concordant with our code of
conduct.
We strongly recommend adhering to Bioconductor’s coding
guide for
software consistency if you wish to contribute to mixOmics R codes.
To report a bug (or offer a solution for a bug!) visit: https://github.com/mixOmicsTeam/mixOmics/issues. We fully welcome and appreciate well-formatted and detailed pull requests. Preferably with tests on our datasets.
We wish to make our discussions transparent so please direct your analysis questions to our discussion forum https://mixomics-users.discourse.group. This forum is aimed to host discussions on choices of multivariate analyses, as well as comments and suggestions to improve the package. We hope to create an active community of users, data analysts, developers and R programmers alike! Thank you!
mixOmics is collaborative project between Australia (Melbourne),
France (Toulouse), and Canada (Vancouver). The core team includes
Kim-Anh Lê Cao - https://lecao-lab.science.unimelb.edu.au (University
of Melbourne), Florian Rohart - http://florian.rohart.free.fr
(Toulouse) and Sébastien Déjean -
https://perso.math.univ-toulouse.fr/dejean/. We also have key
contributors, past (Benoît Gautier, François Bartolo) and present (Al
Abadi, University of Melbourne) and several collaborators including
Amrit Singh (University of British Columbia), Olivier Chapleur (IRSTEA,
Paris), Antoine Bodein (Universite de Laval) - it could be you too, if
you wish to be involved!.
The project started at the Institut de Mathématiques de Toulouse in
France, and has been fully implemented in Australia, at the University
of Queensland, Brisbane (2009 – 2016) and at the University of
Melbourne, Australia (from 2017). We focus on the development of
computational and statistical methods for biological data integration
and their implementation in mixOmics.
mixOmics offers a wide range of novel multivariate methods for the
exploration and integration of biological datasets with a particular
focus on variable selection. Single ‘omics analysis does not provide
enough information to give a deep understanding of a biological system,
but we can obtain a more holistic view of a system by combining multiple
‘omics analyses. Our mixOmics R package proposes a whole range of
multivariate methods that we developed and validated on many biological
studies to gain more insight into ‘omics biological studies.
www.mixOmics.org (tutorials and resources)
Our latest bookdown vignette: https://mixomicsteam.github.io/Bookdown/.
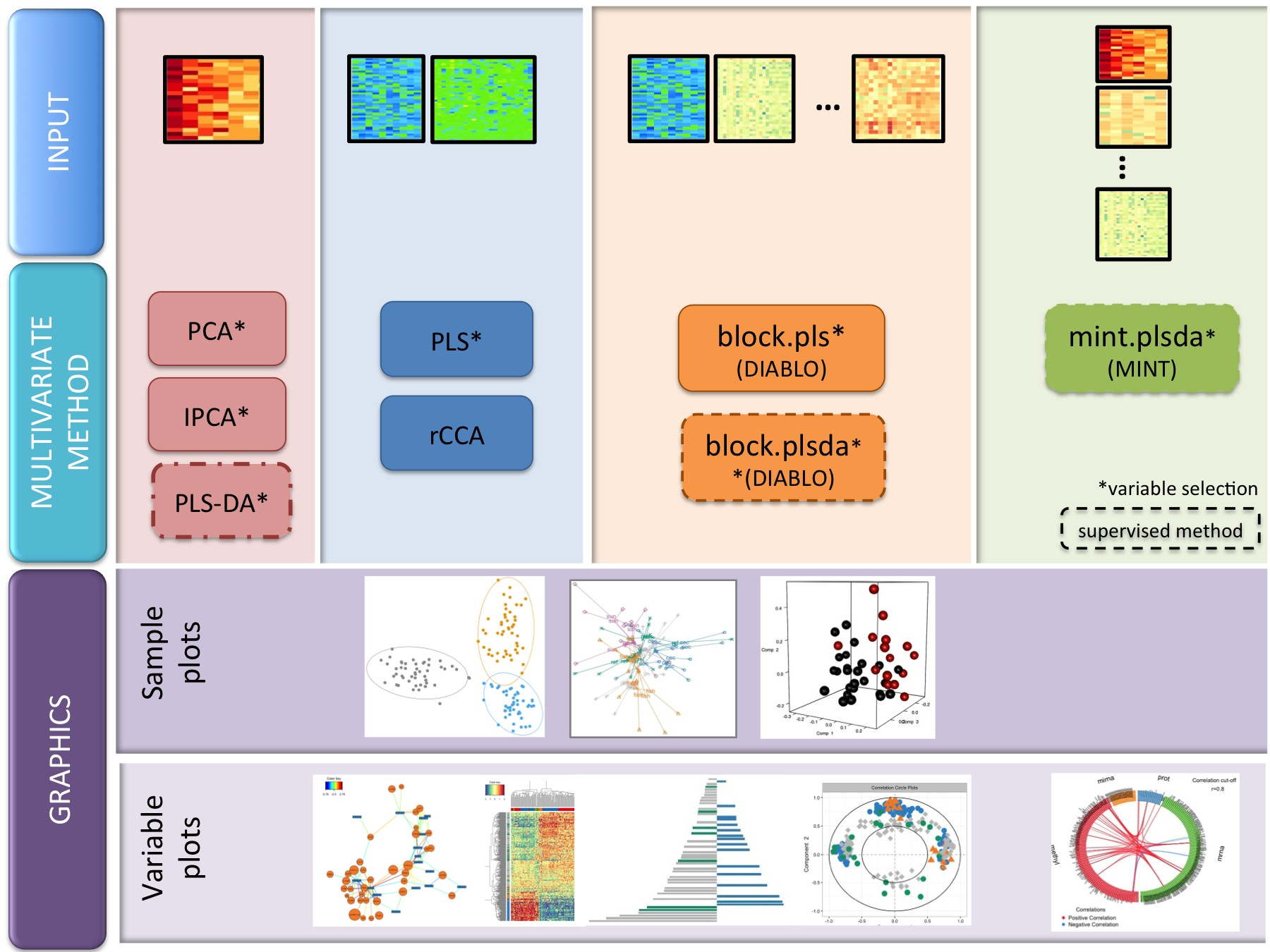
We have developed 17 novel multivariate methods (the package includes 19 methods in total). The names are full of acronyms, but are represented in this diagram. PLS stands for Projection to Latent Structures (also called Partial Least Squares, but not our preferred nomenclature), CCA for Canonical Correlation Analysis.
That’s it! Ready! Set! Go!
Thank you for using mixOmics!
- New biplot now available for
pcafamily. See the examples in this issue
- weighted consensus plots for DIABLO objects now consider per-component weights
-
plotIndivnow supports (weighted) consensus plots for block analyses. See the example in this issue -
plotIndiv(..., ind.names=FALSE)warning issue now fixed
perf.block.splsdanow supports calculation of combined AUCblock.splsdabug which could drop some classes withnear.zero.variance=TRUEnow fixed
-
Parallel computing improved for
tuneandperffunctions, and even more on Unix-like systems -
Fixed margin error problem with
plotLoadings. See the example in this issue -
cimbug which overestimated correlations for single component now fixed -
perf.sgccdanow supports calculation of average combined AUC
- You can now customise
aurocplots in version 6.8.3. See example here