Tuxedo Genome Guided Transcriptome Assembly Workshop
The following details the steps involved in:
- Aligning RNA-Seq reads to a genome using Tophat
- Assembling transcript structures from read alignments using Cufflinks
- Visualizing reads and transcript structures using IGV
- Performing differential expression analysis using Cuffdiff
- Expression analysis using CummeRbund
All required software and data are provided pre-installed on a VirtualBox image. Be sure to run the workshop VM and open a terminal.
After installing the VM, be sure to quickly update the contents of the rnaseq_workshop_data directory by:
% cd RNASeq_Trinity_Tuxedo_Workshop
% git pull
This way, you’ll have the latest content, including any recent bugfixes.
This demo uses RNA-Seq data corresponding to Schizosaccharomyces pombe (fission yeast), involving paired-end 76 base strand-specific RNA-Seq reads corresponding to four samples: Sp_log (logarithmic growth), Sp_plat (plateau phase), Sp_hs (heat shock), and Sp_ds (diauxic shift).
There are 'left.fq' and 'right.fq' FASTQ formatted Illlumina read files for each of the four samples. All RNA-Seq data sets can be found in the RNASEQ_data/ subdirectory.
Also included is a 'genome.fa' file corresponding to a genome sequence, and annotations for reference genes ('genes.bed' or 'genes.gff3'). These resources can be found in the GENOME_data/ subdirectory.
Note, although the genes, annotations, and reads represent genuine sequence data, they were artificially selected and organized for use in this tutorial, so as to provide varied levels of expression in a very small data set, which could be processed and analyzed within an approximately one hour time session and with minimal computing resources.
The commands to be executed are indicated below after a command prompt '%'. You can type these commands into the terminal, or you can simply copy/paste these commands into the terminal to execute the operations.
To avoid having to copy/paste the numerous commands shown below into a unix terminal, the VM includes a script ‘runTrinityDemo.pl’ that enables you to run each of the steps interactively. To begin, simply run:
% ./runTuxedoDemo.pl
Note, by default and for convenience, the demo will show you the commands that are to be executed. This way, you don’t need to type them in yourself.
The protocol followed below is that described in
and follows this general framework as illustrated in the above publication:
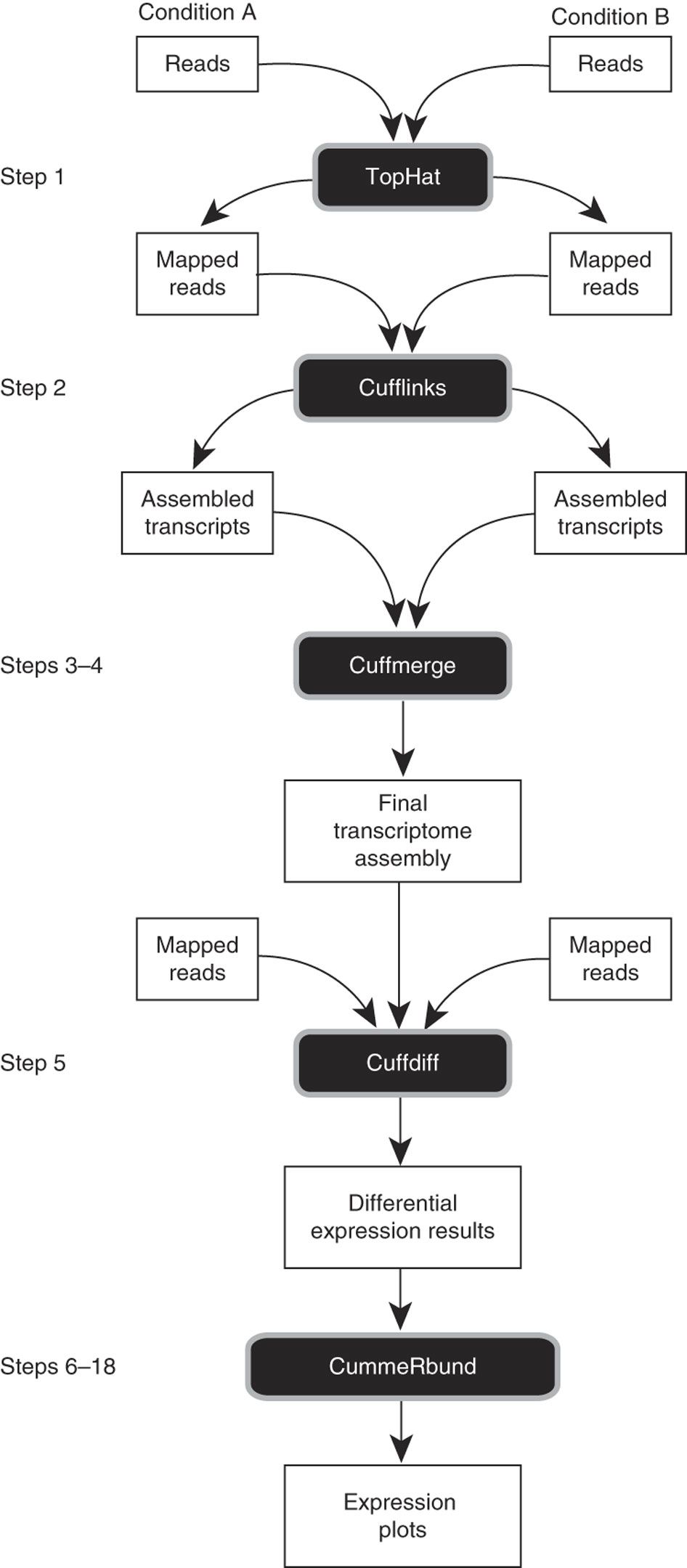
First, prepare the ‘genome.fa’ file for tophat alignment:
% bowtie2-build GENOME_data/genome.fa genome
Align reads using tophat:
% tophat2 -I 1000 -i 20 --library-type fr-firststrand \
-o tophat.Sp_ds.dir genome \
RNASEQ_data/Sp_ds.left.fq.gz RNASEQ_data/Sp_ds.right.fq.gz
Rename the alignment (bam) output file according to this sample name:
% mv tophat.Sp_ds.dir/accepted_hits.bam tophat.Sp_ds.dir/Sp_ds.bam
Index this bam file for later viewing using IGV:
% samtools index tophat.Sp_ds.dir/Sp_ds.bam
Reconstruct transcripts for this sample using Cufflinks:
% cufflinks --no-update-check --overlap-radius 1 \
--library-type fr-firststrand \
-o cufflinks.Sp_ds.dir tophat.Sp_ds.dir/Sp_ds.bam
Rename the cufflinks transcript structure output file according to this sample:
% mv cufflinks.Sp_ds.dir/transcripts.gtf cufflinks.Sp_ds.dir/Sp_ds.transcripts.gtf
Now, you’re done with running Tuxedo on this sample. You now need to repeat these operations for each of the three other samples, as below:
% tophat2 -I 1000 -i 20 --library-type fr-firststrand \
-o tophat.Sp_hs.dir genome \
RNASEQ_data/Sp_hs.left.fq.gz RNASEQ_data/Sp_hs.right.fq.gz
% mv tophat.Sp_hs.dir/accepted_hits.bam tophat.Sp_hs.dir/Sp_hs.bam
% samtools index tophat.Sp_hs.dir/Sp_hs.bam
% cufflinks --no-update-check --overlap-radius 1 \
--library-type fr-firststrand \
-o cufflinks.Sp_hs.dir tophat.Sp_hs.dir/Sp_hs.bam
% mv cufflinks.Sp_hs.dir/transcripts.gtf cufflinks.Sp_hs.dir/Sp_hs.transcripts.gtf
% tophat2 -I 1000 -i 20 --library-type fr-firststrand \
-o tophat.Sp_log.dir genome \
RNASEQ_data/Sp_log.left.fq.gz RNASEQ_data/Sp_log.right.fq.gz
% mv tophat.Sp_log.dir/accepted_hits.bam tophat.Sp_log.dir/Sp_log.bam
% samtools index tophat.Sp_log.dir/Sp_log.bam
% cufflinks --no-update-check --overlap-radius 1 \
--library-type fr-firststrand \
-o cufflinks.Sp_log.dir tophat.Sp_log.dir/Sp_log.bam
% mv cufflinks.Sp_log.dir/transcripts.gtf cufflinks.Sp_log.dir/Sp_log.transcripts.gtf
% tophat2 -I 1000 -i 20 --library-type fr-firststrand \
-o tophat.Sp_plat.dir genome \
RNASEQ_data/Sp_plat.left.fq.gz RNASEQ_data/Sp_plat.right.fq.gz
% mv tophat.Sp_plat.dir/accepted_hits.bam tophat.Sp_plat.dir/Sp_plat.bam
% samtools index tophat.Sp_plat.dir/Sp_plat.bam
% cufflinks --no-update-check --overlap-radius 1 \
--library-type fr-firststrand \
-o cufflinks.Sp_plat.dir tophat.Sp_plat.dir/Sp_plat.bam
% mv cufflinks.Sp_plat.dir/transcripts.gtf cufflinks.Sp_plat.dir/Sp_plat.transcripts.gtf
First, create a file that lists the names of the files containing the separately reconstructed transcripts, which can be done like so, writing each of the four Cufflinks transcript GTF files to a newly created ‘assemblies.txt’ file.
% echo cufflinks.Sp_ds.dir/Sp_ds.transcripts.gtf > assemblies.txt
% echo cufflinks.Sp_hs.dir/Sp_hs.transcripts.gtf >> assemblies.txt
% echo cufflinks.Sp_log.dir/Sp_log.transcripts.gtf >> assemblies.txt
% echo cufflinks.Sp_plat.dir/Sp_plat.transcripts.gtf >> assemblies.txt
Verify that this file now contains all gtf filenames to be included in the merge:
% cat assemblies.txt
.
cufflinks.Sp_ds.dir/Sp_ds.transcripts.gtf
cufflinks.Sp_hs.dir/Sp_hs.transcripts.gtf
cufflinks.Sp_log.dir/Sp_log.transcripts.gtf
cufflinks.Sp_plat.dir/Sp_plat.transcripts.gtf
And now we’re ready to merge the transcripts using cuffmerge:
% cuffmerge -s GENOME_data/genome.fa assemblies.txt
The merged set of transcripts should now exist as file 'merged_asm/merged.gtf'.
View the reconstructed transcripts and the tophat alignments in IGV
% igv.sh -g `pwd`/GENOME_data/genome.fa \
`pwd`/merged_asm/merged.gtf,`pwd`/GENOME_data/genes.bed,`pwd`/tophat.Sp_ds.dir/Sp_ds.bam,`pwd`/tophat.Sp_hs.dir/Sp_hs.bam,`pwd`/tophat.Sp_log.dir/Sp_log.bam,`pwd`/tophat.Sp_plat.dir/Sp_plat.bam
Note, you may need to resize the individual alignment patterns in the viewer by dragging the panel boundaries. Afterwards, go to menu “Tracks” -> “Fit Data To Window” to re-space the contents of the viewer)

Pan the genome, examine the alignments, known genes and reconstructed genes, and contemplate the following:
-
Do the alignments agree with the known gene structures (ex. intron placements)?
-
Do the cufflinks-reconstructed transcripts well represent the alignments?
-
Do the cufflinks-reconstructed transcripts match the structures of the known transcripts?
% cuffdiff --no-update-check --library-type fr-firststrand \
-o diff_out -b GENOME_data/genome.fa \
-L Sp_ds,Sp_hs,Sp_log,Sp_plat \
-u merged_asm/merged.gtf \
tophat.Sp_ds.dir/Sp_ds.bam \
tophat.Sp_hs.dir/Sp_hs.bam \
tophat.Sp_log.dir/Sp_log.bam \
tophat.Sp_plat.dir/Sp_plat.bam
Examine the output files generated in the diff_out/ directory.
A table containing the results from the gene-level differential expression analysis can be found as ‘diff_out/gene_exp.diff’. Examine the top lines of this file like so:
% head diff_out/gene_exp.diff
.
test_id gene_id gene locus sample_1 sample_2 status value_1 value_2 log2(fold_change) test_stat p_value q_value significant
XLOC_000001 XLOC_000001 - genome:125-2354 Sp_ds Sp_hs NOTEST 95.9855 92.9767 -0.0459471 0 1 1 no
XLOC_000002 XLOC_000002 - genome:5711-7839 Sp_ds Sp_hs OK 292.858 331.841 0.180291 0.211743 0.851 0.986587 no
XLOC_000003 XLOC_000003 - genome:8067-8895 Sp_ds Sp_hs OK 2367.86 1226.53 -0.949004 -1.5155 0.3499 0.835113 no
XLOC_000004 XLOC_000004 - genome:15162-15953 Sp_ds Sp_hs OK 146.177 177.165 0.277377 0.317119 0.84325 0.986587 no
XLOC_000005 XLOC_000005 - genome:16257-17233 Sp_ds Sp_hs OK 1595.2 1745.41 0.129826 0.157899 0.9053 0.986587 no
XLOC_000006 XLOC_000006 - genome:18775-19820 Sp_ds Sp_hs OK 164.828 856.768 2.37794 2.97705 0.0385 0.440712 no
XLOC_000007 XLOC_000007 - genome:20136-21633 Sp_ds Sp_hs OK 352.268 185.928 -0.921934 -1.27306 0.32635 0.831724 no
XLOC_000008 XLOC_000008 - genome:23642-24694 Sp_ds Sp_hs OK 20006.1 378.726 -5.72314 -4.42028 0.0215 0.31522 no
XLOC_000009 XLOC_000009 - genome:30960-32340 Sp_ds Sp_hs OK 234.796 229.581 -0.0324032 -0.0538314 0.9638 0.990617 no
The Tuxedo software suite includes the cummeRbund tool for data exploration. cummeRbund is an R based software tool included in Bioconductor.
Use ‘cummeRbund’ to analyze the results from cuffdiff like so. First, begin an R session:
% R
note, to exit R, type cntrl-D, or type “q()” ).
Load the cummerbund library into the R session
> library(cummeRbund)
Import the cuffdiff results
> cuff = readCufflinks('diff_out')
> csDensity(genes(cuff))

Expression values are typically log-normally distributed. This is just a sanity check.
> csScatter(genes(cuff), 'Sp_log', 'Sp_plat')

Strongly differentially expressed transcripts should fall far from the linear regression line.
Examine individual sample distributions of gene expression values and the pairwise scatterplots together in a single plot.
> csScatterMatrix(genes(cuff))

> csVolcanoMatrix(genes(cuff))

Retrieve the gene-level differential expression data
> gene_diff_data = diffData(genes(cuff))
How many ‘genes’ are there?
> nrow(gene_diff_data)
.
1248
From the gene-level differential expression data, extract those that are labeled as significantly different.
note, normally just set criteria as “significant=’yes’”, but we’re adding an additional p_value filter just to capture some additional transcripts for demonstration purposes only. This simulated data is overly sparse and actually suboptimal for this demonstration (in hindsight).
> sig_gene_data = subset(gene_diff_data,(significant=='yes' | p_value < 0.01))
How many genes are significantly DE according to these criteria?
> nrow(sig_gene_data)
.
40
> head(sig_gene_data)
.
gene_id sample_1 sample_2 status value_1 value_2 log2_fold_change
57 XLOC_000057 Sp_ds Sp_hs OK 33316.60 116.3020 -8.16222
137 XLOC_000137 Sp_ds Sp_hs OK 30059.50 245.2130 -6.93764
147 XLOC_000147 Sp_ds Sp_hs OK 1121.29 70.6636 -3.98804
245 XLOC_000031 Sp_ds Sp_log OK 19082.60 351.1190 -5.76415
271 XLOC_000057 Sp_ds Sp_log OK 33316.60 109.1140 -8.25427
288 XLOC_000074 Sp_ds Sp_log OK 0.00 570.1320 Inf
test_stat p_value q_value significant
57 -4.86285 0.00510 0.255680 no
137 -4.45927 0.00655 0.255680 no
147 -4.02931 0.00590 0.255680 no
245 -7.05283 0.00830 0.255680 no
271 -4.77024 0.00500 0.255680 no
288 NA 0.00195 0.140171 no
You can write the list of significantly differentially expressed genes to a file like so:
> write.table(sig_gene_data, 'sig_diff_genes.txt', sep = '\t', quote = F)
# verify it worked:
> system("head sig_diff_genes.txt") # run unix command from within R
Select expression info for the one gene by its gene identifier: Let’s take the first gene identifier in our sig_gene_data table:
# first, get its gene_id
> ex_gene_id = sig_gene_data$gene_id[1]
# print its value to the screen:
> ex_gene_id
.
"XLOC_000057"
Get that gene ‘object’ from cummeRbund and assign it to variable ‘ex_gene’
> ex_gene = getGene(cuff, ex_gene_id)
Now plot the expression values for the gene under each condition
Note, error bars are only turned off here because this data set is both simulated and hugely underpowered to have reasonable confidence levels
> expressionBarplot(ex_gene, logMode=T, showErrorbars=F)

First retrieve the ‘genes’ from the ‘cuff’ data set by providing a list of gene identifiers like so:
> sig_genes = getGenes(cuff, sig_gene_data$gene_id)
Now draw the heatmap
> csHeatmap(sig_genes, cluster='both')
 /
/
And that's it for our introduction to Tuxedo. More information on using the Tuxedo package can be found at:
-
The Tuxedo Nature Protocol paper: [Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012 Mar 1;7(3):562-78. doi: 10.1038/nprot.2012.016.] (http://www.nature.com/nprot/journal/v7/n3/full/nprot.2012.016.html)
-
The CummeRbund manual: http://compbio.mit.edu/cummeRbund/manual_2_0.html
Note, most of the tutorial provided here is based on the above two resources)
and the Tuxedo tool websites:
- TopHat: http://ccb.jhu.edu/software/tophat/index.shtml
- Cufflinks: http://cole-trapnell-lab.github.io/cufflinks/
- CummeRbund: http://compbio.mit.edu/cummeRbund/