Python package
MetaLogo provides stand alone package for user to draw figures in their own computer or server. There are two ways to make sequence logos using MetaLogo package. One is importing MetaLogo into your python scripts and create logos with specific parameters, the other is directly execute MetaLogo in system terminal and pass arguments into MetaLogo to custom the logos. These two ways share the same set of parameters, which we will explain in this tutorial.
When you installed MetaLogo, you can run MetaLogo in your terminal like this:
$ metalogo --seq_file MetaLogo/examples/example.fa --output_dir . --output_name test.png --withtree
If you do not want to install MetaLogo as a package in your system, you can also directly run MetaLogo like below:
$ python -m MetaLogo.MetaLogo.entry --seq_file MetaLogo/examples/example.fa --output_dir . --output_name test.png --withtree
Note the current workdir is the directory where MetaLogo source code exists.
Make sure you have install all the requirements and be under MetaLogo project directory.
If the command run successfully, you will get a plot named test.png in your current directory.

Below are the parameters you can pass into MetaLogo.
usage: metalogo [-h] [--config CONFIG]
[--type {Horizontal,Circle,Radiation,Threed}]
[--seq_file SEQ_FILE] [--seq_file_type {fasta,fastq}]
[--sequence_type {auto,dna,rna,aa}] [--task_name TASK_NAME]
[--min_length MIN_LENGTH] [--max_length MAX_LENGTH]
[--group_strategy {auto,length,identifier}]
[--clustering_method CLUSTERING_METHOD]
[--group_resolution GROUP_RESOLUTION]
[--group_limit GROUP_LIMIT]
[--group_order {length,length_reverse,identifier,identifier_reverse}]
[--color_scheme {basic_dna_color,basic_rna_color,basic_aa_color}]
[--color_scheme_json_str COLOR_SCHEME_JSON_STR]
[--color_scheme_json_file COLOR_SCHEME_JSON_FILE]
[--height_algorithm {bits,bits_without_correction,probabilities}]
[--align] [--padding_align]
[--align_metric {dot_product,js_divergence,cosine,entropy_bhattacharyya}]
[--connect_threshold CONNECT_THRESHOLD]
[--gap_score GAP_SCORE]
[--display_range_left DISPLAY_RANGE_LEFT]
[--display_range_right DISPLAY_RANGE_RIGHT] [--withtree]
[--logo_margin_ratio LOGO_MARGIN_RATIO]
[--column_margin_ratio COLUMN_MARGIN_RATIO]
[--char_margin_ratio CHAR_MARGIN_RATIO] [--hide_version_tag]
[--hide_left_axis] [--hide_right_axis] [--hide_top_axis]
[--hide_bottom_axis] [--hide_x_ticks] [--hide_y_ticks]
[--hide_z_ticks] [--x_label X_LABEL] [--y_label Y_LABEL]
[--z_label Z_LABEL] [--show_group_id] [--show_grid]
[--title_size TITLE_SIZE] [--label_size LABEL_SIZE]
[--tick_size TICK_SIZE] [--group_id_size GROUP_ID_SIZE]
[--figure_size_x FIGURE_SIZE_X]
[--figure_size_y FIGURE_SIZE_Y] [--auto_size]
[--align_color ALIGN_COLOR] [--align_alpha ALIGN_ALPHA]
[--output_dir OUTPUT_DIR] [--output_name OUTPUT_NAME]
[--fa_output_dir FA_OUTPUT_DIR] [--uid UID]
[--logo_format {png,pdf}] [--analysis]
[--clustalo_bin CLUSTALO_BIN] [--fasttree_bin FASTTREE_BIN]
[--fasttreemp_bin FASTTREEMP_BIN]
[--treecluster_bin TREECLUSTER_BIN] [-v]
optional arguments:
-h, --help show this help message and exit
--config CONFIG The config file contain sequences (default: None)
--type {Horizontal,Circle,Radiation,Threed}
Choose the layout type of sequence logo (default: Horizontal)
--seq_file SEQ_FILE The input file contain sequences (default: None)
--seq_file_type {fasta,fastq}
The type of input file (default: fasta)
--sequence_type {auto,dna,rna,aa}
The type of sequences (default: auto)
--task_name TASK_NAME
The title to displayed on the figure (default: MetaLogo)
--min_length MIN_LENGTH
The minimum length of sequences to be included (default: 8)
--max_length MAX_LENGTH
The maximum length of sequences to be included (default: 20)
--group_strategy {auto,length,identifier}
The strategy to separate sequences into groups (default: auto)
--clustering_method CLUSTERING_METHOD
The method for tree clustering (default: max)
--group_resolution GROUP_RESOLUTION
The resolution for sequence grouping (default: 0.5)
--group_limit GROUP_LIMIT
The limit for group number (default: 20)
--group_order {length,length_reverse,identifier,identifier_reverse}
The order of groups (default: length)
--color_scheme {basic_dna_color,basic_rna_color,basic_aa_color}
The color scheme (default: basic_dna_color)
--color_scheme_json_str COLOR_SCHEME_JSON_STR
The json string of color scheme (default: None)
--color_scheme_json_file COLOR_SCHEME_JSON_FILE
The json file of color scheme (default: None)
--height_algorithm {bits,bits_without_correction,probabilities}
The algorithm for character height (default: bits)
--align If show alignment of adjacent sequence logo (default: False)
--padding_align If padding logos to make multiple logo alignment (default: False)
--align_metric {dot_product,js_divergence,cosine,entropy_bhattacharyya}
The metric for align score (default: dot_product)
--connect_threshold CONNECT_THRESHOLD
The align threshold (default: 0.8)
--gap_score GAP_SCORE
The gap score for alignment (default: -1.0)
--display_range_left DISPLAY_RANGE_LEFT
The start position of display range (Global alignment with padding required) (default: 0)
--display_range_right DISPLAY_RANGE_RIGHT
Then end position of display range (Global alignment with padding requirement) (default: -1)
--withtree If show tree besides sequence logo (default: False)
--logo_margin_ratio LOGO_MARGIN_RATIO
Margin ratio between the logos (default: 0.1)
--column_margin_ratio COLUMN_MARGIN_RATIO
Margin ratio between the columns (default: 0.05)
--char_margin_ratio CHAR_MARGIN_RATIO
Margin ratio between the chars (default: 0.05)
--hide_version_tag If show version tag of MetaLogo (default: False)
--hide_left_axis If hide left axis (default: False)
--hide_right_axis If hide right axis (default: False)
--hide_top_axis If hide top axis (default: False)
--hide_bottom_axis If hide bottom axis (default: False)
--hide_x_ticks If hide ticks of X axis (default: False)
--hide_y_ticks If hide ticks of Y axis (default: False)
--hide_z_ticks If hide ticks of Z axis (default: False)
--x_label X_LABEL The label for X axis (default: None)
--y_label Y_LABEL The label for Y axis (default: None)
--z_label Z_LABEL The label for Z axis (default: None)
--show_group_id If show group ids (default: False)
--show_grid If show background grid (default: False)
--title_size TITLE_SIZE
The size of figure title (default: 20)
--label_size LABEL_SIZE
The size of figure xy labels (default: 10)
--tick_size TICK_SIZE
The size of figure ticks (default: 10)
--group_id_size GROUP_ID_SIZE
The size of group labels (default: 10)
--figure_size_x FIGURE_SIZE_X
The width of figure (default: 20)
--figure_size_y FIGURE_SIZE_Y
The height of figure (default: 10)
--auto_size Let MetaLogo determine the size of figures (default: False)
--align_color ALIGN_COLOR
The color of alignment (default: blue)
--align_alpha ALIGN_ALPHA
The transparency of alignment (default: 0.2)
--output_dir OUTPUT_DIR
Output path of figure (default: figure_output)
--output_name OUTPUT_NAME
Output name of figure (default: test.png)
--fa_output_dir FA_OUTPUT_DIR
Output path of fas (default: sequence_input)
--uid UID Task id (default:
a878a8bd-6818-4bf8-91ad-f148d67f849a)
--logo_format {png,pdf}
The format of figures (default: png)
--analysis If perform basic analysis on data (default: False)
--clustalo_bin CLUSTALO_BIN
The path of clustalo bin (default:
dependencies/clustalo)
--fasttree_bin FASTTREE_BIN
The path of fasttree bin (default:
dependencies/FastTree)
--fasttreemp_bin FASTTREEMP_BIN
The path of fasttreeMP bin (default:
dependencies/FastTreeMP)
--treecluster_bin TREECLUSTER_BIN
The path of treecluster bin (default: TreeCluster.py)
-v, --version show program's version number and exit
Most of the parameters are easy to understand, there are several parameters need to be explained here.
--group_strategy {auto,length,identifier}
The strategy to separate sequences into groups
(default: auto)
This parameter specify the way you group sequences. In default, MetaLogo groups sequences by phylogenetic tree. Multiple sequence alignment and phylogenetic tree construction will be automatically performed to cluster the sequences.
However, you could still group sequences by other strategy. MetaLogo can identify group information of sequences from their sequence name. Blow is a example:
>seq1 group@1-fisrtgroup
AATATACAGATACCCATAC
>seq2 group@2-secondgroup
ATACAATACCCACAGATAC
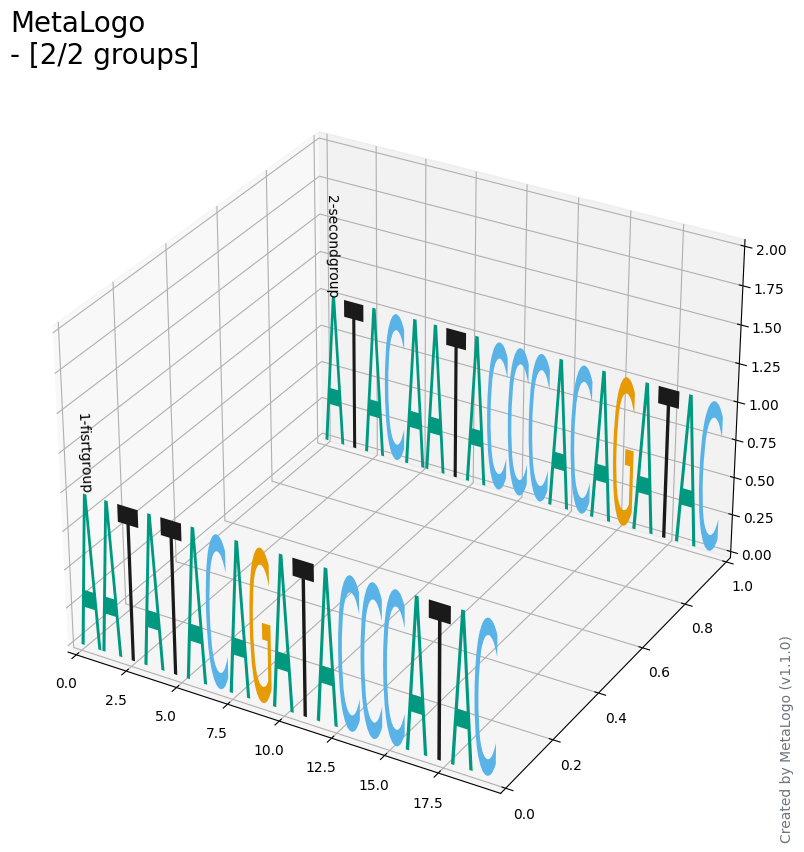
You need to add a 'group@\d-\S' pattern in your sequence names. In the term, 'group@' is fixed and then followed by a number, a dash and a string to indicate group information. Then if you set --group_strategy as 'identifier', MetaLogo will draw sequence logos for different groups. It should be noted that in each group, lengths of sequences must be the same. Below is a output of identifier-grouped input (probabilities as height, 3D layout):
$cat test.fa
>seq1 group@1-fisrtgroup
AATATACAGATACCCATAC
>seq2 group@2-secondgroup
ATACAATACCCACAGATAC
$metalogo --seq_file test.fa --output_dir . --output_name test.png --height_algorithm probabilities --group_strategy identifier --type Threed --show_group_id
--group_order {auto,length,length_reverse,identifier,identifier_reverse}
The order of groups (default: auto)
This parameter specify how to order the groups. 'auto' means automatically sorting groups. 'length' means sorting groups by sequence lengths, 'length_reverse' means sorting groups by sequence lengths in a decreasing order, 'identifier' means sorting groups by its group id indicated in sequence names, i.e. the number followed 'group@' term in sequence name, 'identifier_reverse' means a decreasing order.
--max_length, --min_length
These two parameters specify the length of sequences to be included in the logo drawing process. Sometimes the length range of sequences could be too large for visualization, users could limit the lengths of sequences for sequence logos.
--color_scheme_json_file
This parameter specify the color scheme json file for sequence logo. There are four built-in schemes, namely basic_dna_color,basic_rna_color,basic_aa_color. User can also pass a json format of a python dict into color scheme. Below is a example:
$ cat color.json
{"A": "red", "T": "blue", "G": "yellow", "C": "green"}
$metalogo --seq_file MetaLogo/examples/ectf.fa --color_scheme_json_file color.json --output_dir .
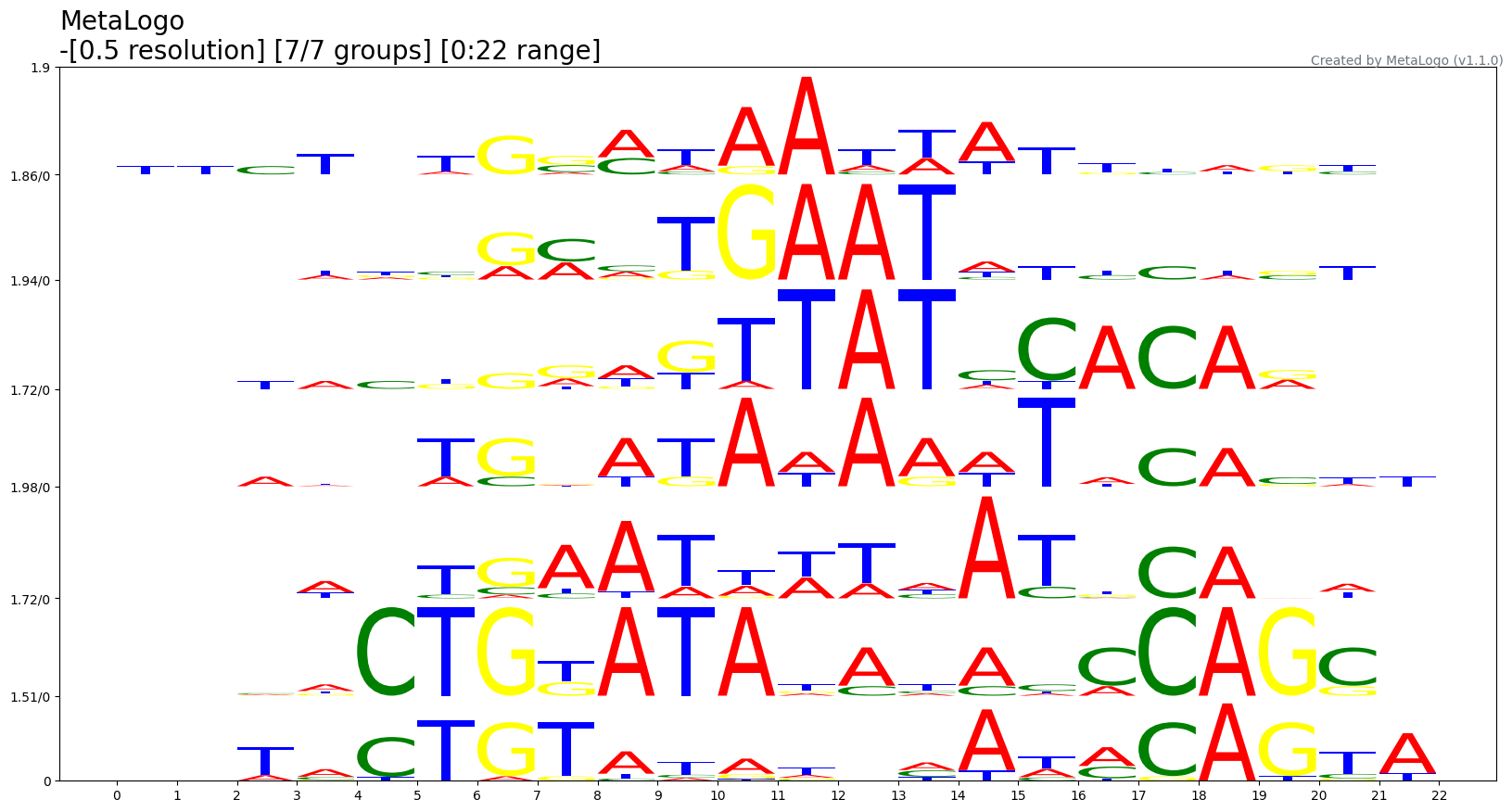
--height_algorithm {bits,probabilities}
The algorithm for character height (default: bits)
This parameter tells MetaLogo to use probabilities or information contents for y axis in sequence logos. If there is only one sequence in one group, the information contents of each positions equal to zeros because error correction. This is the reason why we sometimes use probabilities as height in our tutorial.
$metalogo --seq_file MetaLogo/examples/ectf.fa --output_dir . --height_algorithm probabilities

--align If show alignment of adjacent sequence logo (default: False)
When you pass this parameter to MetaLogo, it will tried to align each pair of groups and highlight the similar positions.
$metalogo --seq_file MetaLogo/examples/ectf.fa --output_dir . --height_algorithm probabilities --align

For the align metric and threshold, you could check the --align_metric and --connect_threshold parameter.
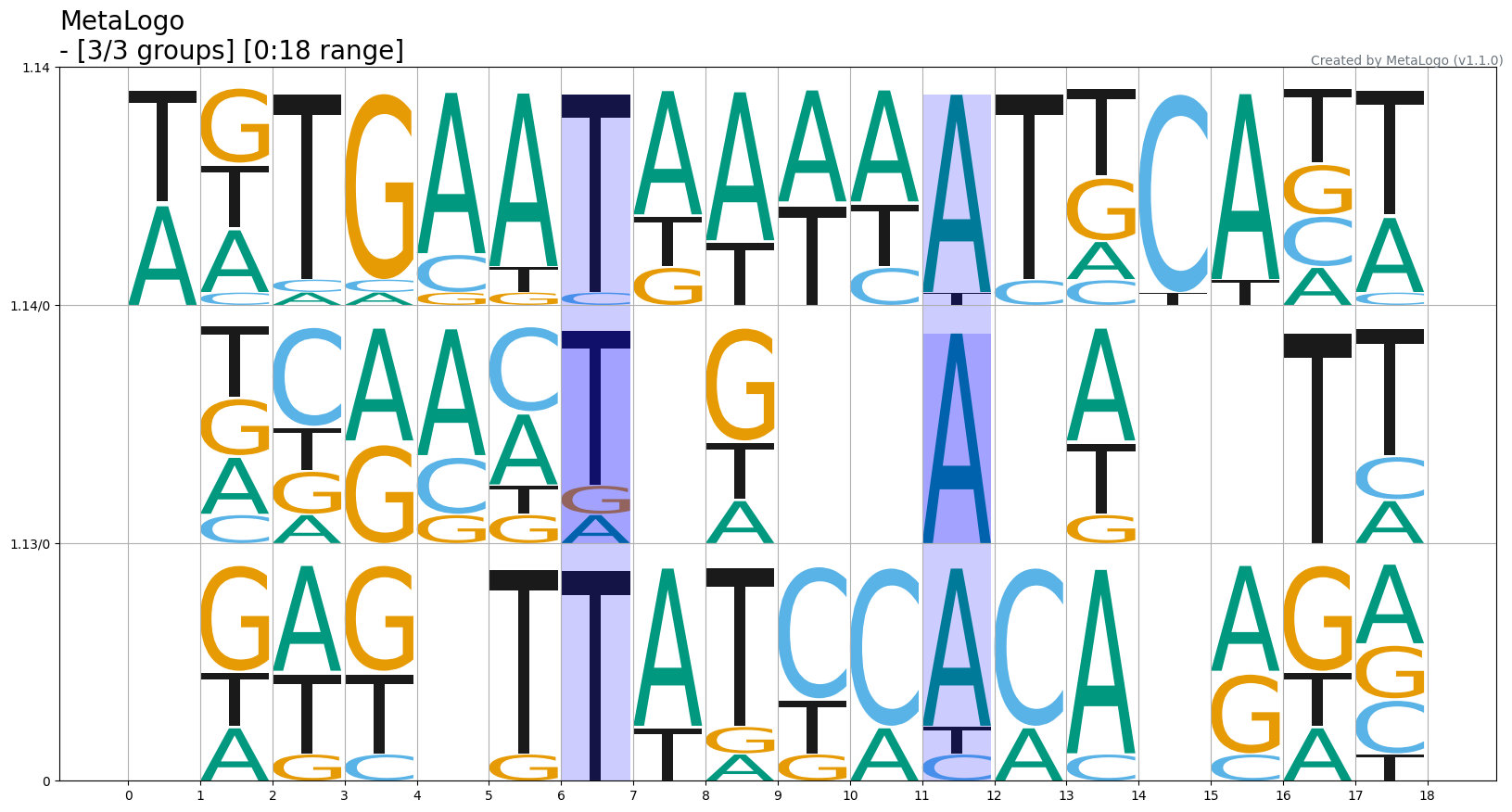
--padding_align If padding logos to make multiple logo alignment
(default: False)
This parameter is only valid for user-defined grouping scenario. If the --group_strategy is set as 'auto', this parameter will not work. In length-grouping or identifier-grouping, this parameter will make MetaLogo perform multiple logo alignment for all the groups rather than only for two adjacent groups.
$metalogo --seq_file MetaLogo/examples/ectf.fa --output_dir . --height_algorithm probabilities --group_strategy length --align --padding_align --show_grid --connect_threshold 0.6
--align_metric {dot_product,js_divergence,cosine,entropy_bhattacharyya}
The metric for align score (default: dot_product)
This parameter specify the algorithm to measure position similarities between sequence logos. Detailed information could be found in our paper.
--connect_threshold CONNECT_THRESHOLD
The align threshold (default: 0.8)
This parameter specify the threshold to connect two positions between two adjacent groups according to logo alignment. If this threshold is positive (>0), MetaLogo will connect two positions if their similarity score is larger than the threshold. If this threshold is negative (>0), MetaLogo will connect two positions if their similarity score is in the top (ratio*100)% of all pairs, in which ratio equals to -1*threshold.
--align_color ALIGN_COLOR
The color of alignment (default: 10)
--align_alpha ALIGN_ALPHA
The transparency of alignment (default: 10)
These two parameter specify the color and transparency of connections between logos.
--analysis If perform basic analysis on data (default: False)
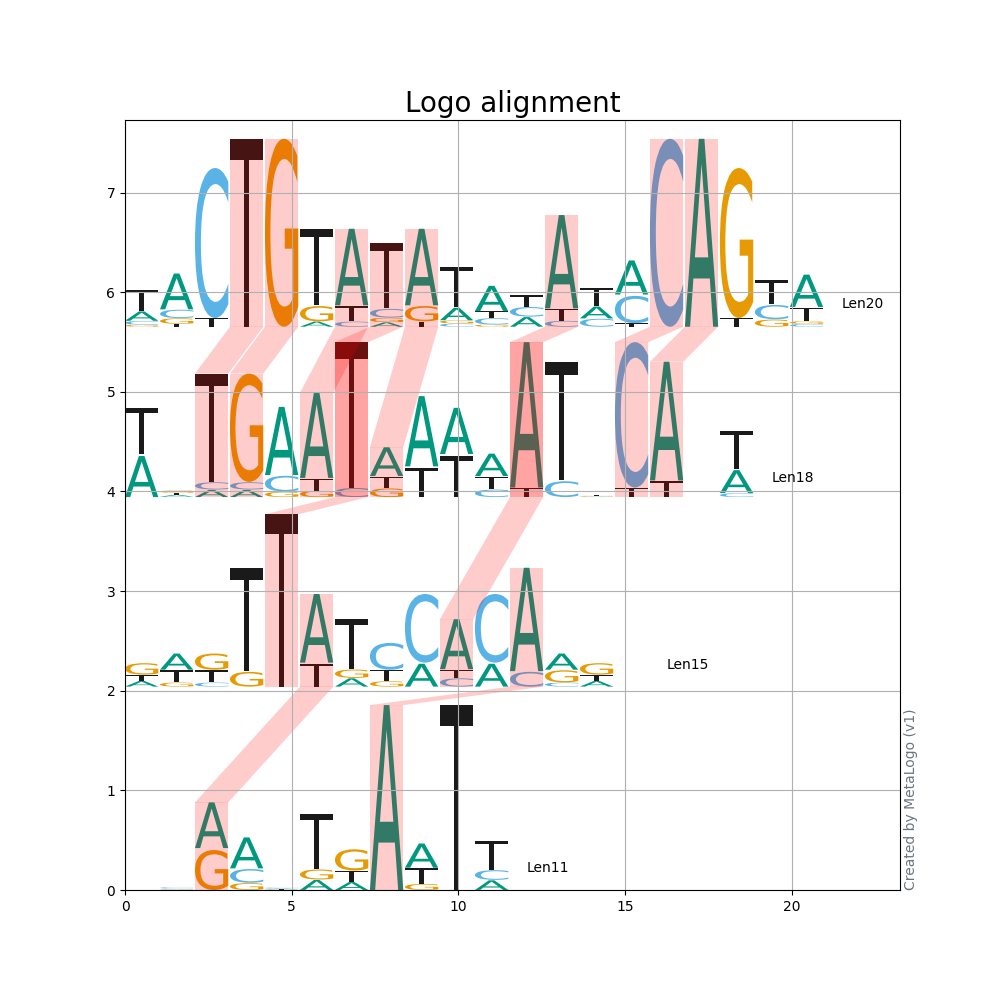
Below is a example for logo alignment.
$ metalogo --input_file examples/ectf.fa --show_group_id --align --padding_align --connect_threshold -0.3 --task_name 'Logo alignment' --show_grid
![]()
Below is a example for logo alignment without global multiple logo alignment and padding.
$ metalogo --input_file examples/ectf.fa --show_group_id --align --connect_threshold -0.3 --task_name 'Logo alignment' --show_grid
--logo_margin_ratio LOGO_MARGIN_RATIO
Margin ratio between the logos (default: 0.1)
--column_margin_ratio COLUMN_MARGIN_RATIO
Margin ratio between the columns (default: 0.05)
--char_margin_ratio CHAR_MARGIN_RATIO
Margin ratio between the chars (default: 0.05)
These three parameters specify the proportional margins between different items.
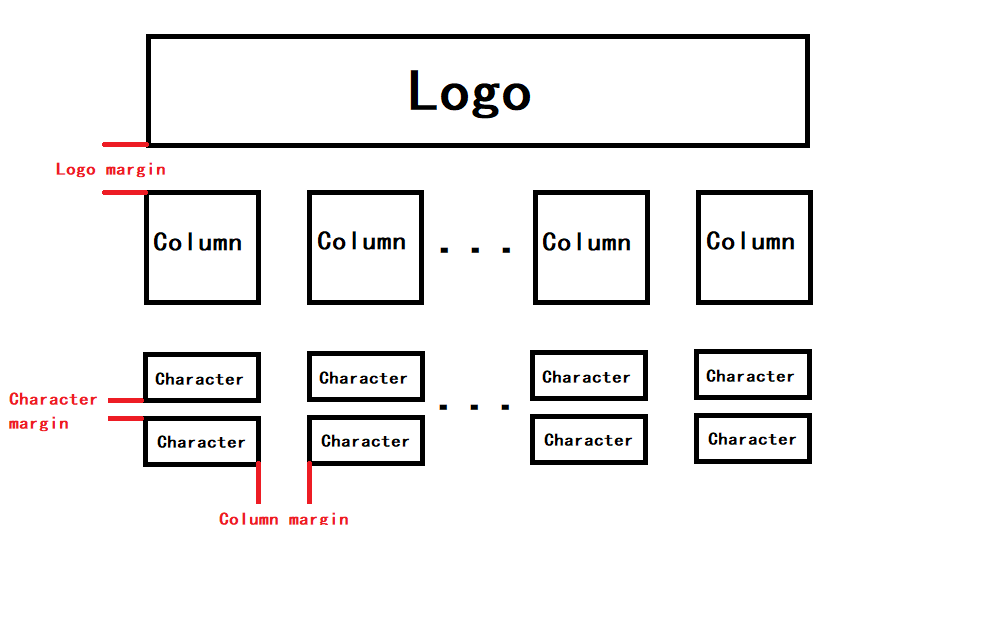
Other parameters are easy to understand according to their names. Most of them are helpful for users to plot custom sequence logos.
If you pass --analysis, MetaLogo will perform basic analysis on the data you input and output related figures in the output directory. Please check the MetaLogo paper or Web Server for details.
MetaLogo will save all the intermediate results, you can specify the path by --fa_output_dir. Files includes:
server.d359d94e-8619-4ff0-8b03-62995a023877.dep.fa #de-duplicated fasta
server.d359d94e-8619-4ff0-8b03-62995a023877.fasttree.cluster # tree clustering result
server.d359d94e-8619-4ff0-8b03-62995a023877.fasttree.rawid.tree #phylogenetic tree with raw sequence name
server.d359d94e-8619-4ff0-8b03-62995a023877.fasttree.tree #phylogenetic tree with new sequence name
server.d359d94e-8619-4ff0-8b03-62995a023877.grouping.fa #grouping details
server.d359d94e-8619-4ff0-8b03-62995a023877.msa.fa #multiple sequence alignment results
server.d359d94e-8619-4ff0-8b03-62995a023877.msa.rawid.fa # multiple sequence alignment results with raw sequence name
server.d359d94e-8619-4ff0-8b03-62995a023877.treedists.csv # sequence distances in the phylogenetic treeAfter install MetaLogo as a python package, you can import MetaLogo into your scripts or notebook easily. Below is a simple example.
from MetaLogo import logo
sequences = [['seq1','ATACAGATACACATCACAG'],['seq2','ATACAGAGATACCAACAGAC'],['seq3','ATACAGAGTTACCCACGGAC']]
bin_args = {
'clustalo_bin':'../MetaLogo/dependencies/clustalo',
'fasttree_bin':'../MetaLogo/dependencies/FastTree',
'fasttreemp_bin':'../MetaLogo/dependencies/FastTreeMP',
}
lg = logo.LogoGroup(sequences,height_algorithm='probabilities',group_strategy='length', **bin_args)
lg.draw()
lg.savefig('test.png')LogoGroup receives nearly same parameters as standalone MetaLogo entry point we described above.
LogoGroup(self, seqs=None, ax=None, group_order='length', group_strategy='length', group_resolution=0.5,
clustering_method = 'max',
start_pos = (0,0), logo_type = 'Horizontal', init_radius=1,
logo_margin_ratio = 0.1, column_margin_ratio = 0.05, char_margin_ratio = 0.05,
align = True, align_metric='sort_consistency', connect_threshold=0.8,
radiation_head_n = 5, threed_interval = 4, color = basic_dna_color, task_name='MetaLogo',
x_label = 'Position', y_label = 'bits',z_label = 'bits', show_grid = True, show_group_id = True,
display_range_left = 0, display_range_right = -1,
hide_left_axis=False, hide_right_axis=False, hide_top_axis=False, hide_bottom_axis=False,
hide_x_ticks=False, hide_y_ticks=False, hide_z_ticks=False,
title_size=20, label_size=10, tick_size=10, group_id_size=10,align_color='blue',align_alpha=0.1,
figure_size_x=-1, figure_size_y=-1,gap_score=-1, padding_align=False, hide_version_tag=False,
sequence_type = 'auto', height_algorithm = 'bits',omit_prob = 0,
seq_file = '', fa_output_dir = '.', output_dir = '.', uid = '',
withtree = False,group_limit=20, target_sequence = '',
clustalo_bin = '', fasttree_bin = '', fasttreemp_bin = '', treecluster_bin = '',
auto_size=True,
*args, **kwargs):For sequences, you need to pass a sequence array into LogoGroup as the first positional parameter. In this sequence array, each item is a tuple of sequence name and its dna or protein sequence. Or you can provide a sequence file with the --seq_file parameter. For color scheme, here you need to pass a python dict into LogoGroup rather than any name string or json formatted dict.
For the structure of MetaLogo, the following figure indicate the class inheritance and method execution order when drawing a MetaLogo.
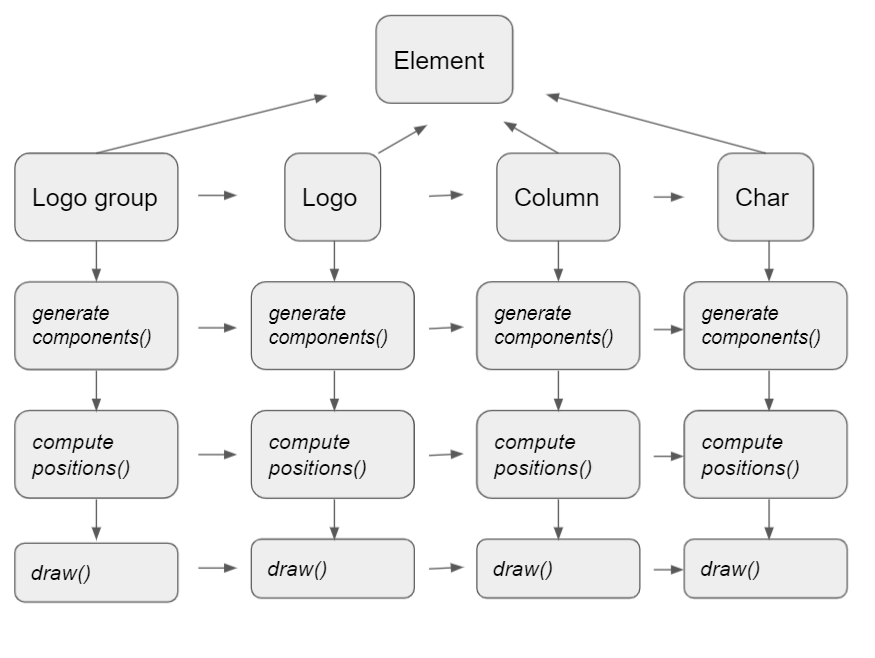
When you using MetaLogo in your project, you could get the ax object of matplotlib as follows:
lg = logo.LogoGroup(sequences,height_algorithm='probabilities',**bin_args)
lg.draw()
ax = logo.axIf you set withtree as True, another matplotlib ax object is also avaliable.
lg = logo.LogoGroup(sequences,withtree=True,**bin_args)
lg.draw()
ax = logo.ax
ax_tree = logo.ax0You could also pass ax to LogoGroup init function when you create LogoGroup instance. Blow is a example.
import matplotlib.pyplot as plt
from MetaLogo import logo
from MetaLogo.colors import basic_dna_color_scheme,basic_aa_color_scheme,basic_rna_color_scheme
sequences = [
['seq1','ATACAGATACACATCACAG'],
['seq2','ATGCAGACACAGATCATAG'],
['seq3','ATACAGAGATACCAACAGAC'],
['seq4','ATACAGAGTTACCCACGGAC'],
['seq5','TTGGAGCGATGCGCCCGGACATC'],
['seq6','TTGGAGCAAAGGCCGCGAATATC'],
['seq7','CTAGAGATGC'],
['seq8','ATAAACAAAC'],
]
ax1 = plt.subplot(221)
ax2 = plt.subplot(222)
ax3 = plt.subplot(223)
ax4 = plt.subplot(224,projection='3d')
paras = {
'height_algrithm':'probabilities',
'padding_align':True,
'task_name':'',
'x_label':'',
'y_label':'',
'z_label':'',
'hide_x_ticks':True,
'hide_y_ticks':True,
'hide_z_ticks':True,
'hide_version_tag':True
}
custom_color = {'A':'red','T':'blue','G':'red','C':'black'}
lg_horizontal = logo.LogoGroup(sequences,logo_type='Horizontal',color=basic_aa_color_scheme, ax=ax1,**paras)
lg_circle = logo.LogoGroup(sequences,logo_type='Circle',ax=ax2,color=basic_dna_color_scheme,**paras)
lg_radiation = logo.LogoGroup(sequences,logo_type='Radiation',color=basic_rna_color_scheme, ax=ax3,**paras)
lg_3d = logo.LogoGroup(sequences,logo_type='Threed',color=custom_color, ax=ax4,**paras)
lg_horizontal.draw()
lg_circle.draw()
lg_radiation.draw()
lg_3d.draw()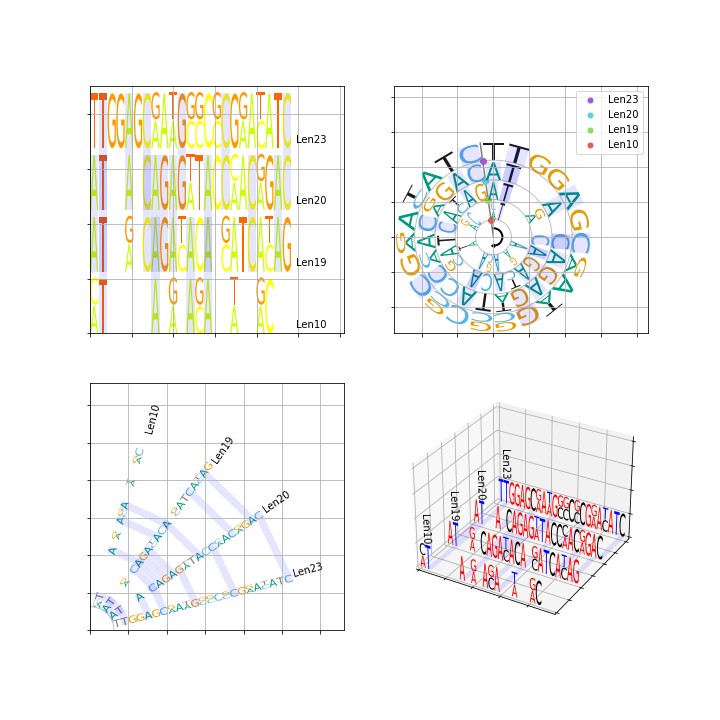
If you want some basic analysis on your data, you could call several functions of MetaLogo to do these stuff. Below are some examples from the entry.py.
fig = logogroup.get_grp_counts_figure().figure
count_name = f'{args.output_dir}/{base_name}.counts.png'
fig.savefig(count_name,bbox_inches='tight')
plt.close(fig)
fig = logogroup.get_seq_lengths_dist().figure
lengths_name = f'{args.output_dir}/{base_name}.lengths.png'
fig.savefig(lengths_name,bbox_inches='tight')
plt.close(fig)
fig = logogroup.get_entropy_figure()
entropy_name = f'{args.output_dir}/{base_name}.entropy.png'
fig.savefig(entropy_name,bbox_inches='tight')
plt.close(fig)
boxplot_entropy_name = f'{args.output_dir}/{base_name}.boxplot_entropy.png'
fig = logogroup.get_boxplot_entropy_figure().figure
fig.savefig(boxplot_entropy_name,bbox_inches='tight')
plt.close(fig)
if args.padding_align or args.group_strategy=='auto':
clustermap_name = f'{args.output_dir}/{base_name}.clustermap.png'
fig = logogroup.get_correlation_figure()
if fig:
fig.savefig(clustermap_name,bbox_inches='tight')
Next: Web Server