This bedtools2 is modified to be complied under cygwin so that it can be used in windows system

Collectively, the bedtools utilities are a swiss-army knife of tools for a wide-range of genomics analysis tasks. The most widely-used tools enable genome arithmetic: that is, set theory on the genome. For example, bedtools allows one to intersect, merge, count, complement, and shuffle genomic intervals from multiple files in widely-used genomic file formats such as BAM, BED, GFF/GTF, VCF.
While each individual tool is designed to do a relatively simple task (e.g., intersect two interval files), quite sophisticated analyses can be conducted by combining multiple bedtools operations on the UNIX command line.
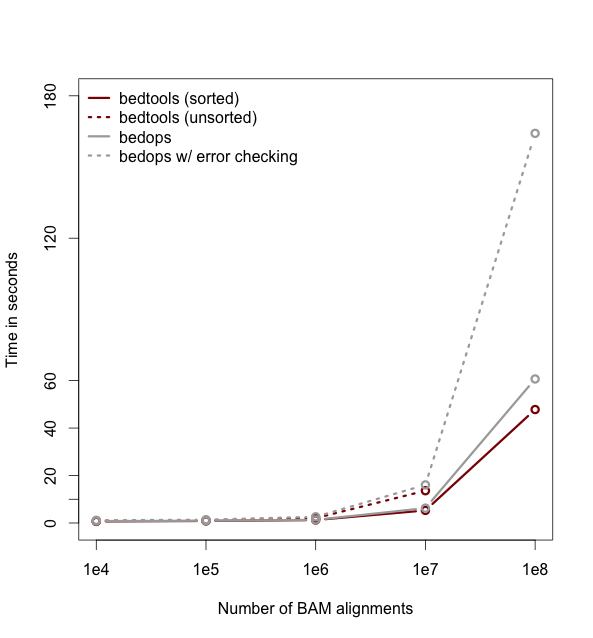
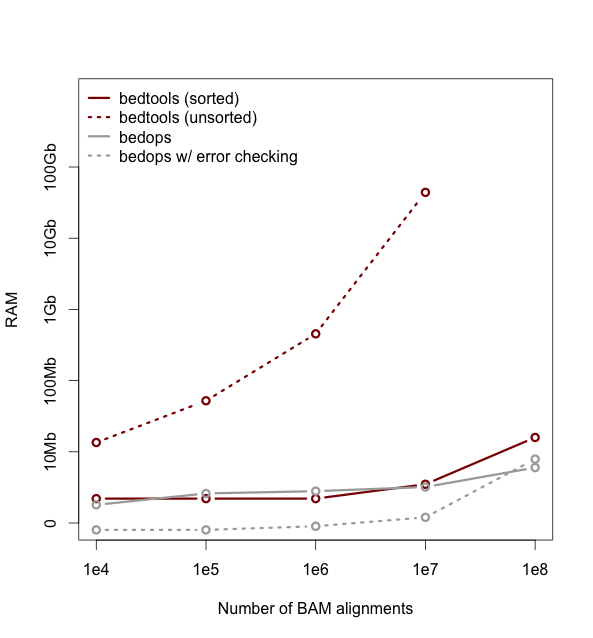
As of version 2.18, bedtools is substantially more scalable thanks to improvements we have made in the algorithm used to process datasets that are pre-sorted
by chromosome and start position. As you can see in the plots below, the speed and memory consumption scale nicely
with sorted data as compared to the poor scaling for unsorted data. The current version of bedtools intersect is as fast as (or slightly faster) than the bedops package's bedmap which uses a similar algorithm for sorted data. The plots below represent counting the number of intersecting alignments from exome capture BAM files against CCDS exons.
The alignments have been converted to BED to facilitate comparisons to bedops. We compare to the bedmap --ec option because similar error checking is enforced by bedtools.
Note: bedtools could not complete when using 100 million alignments and the R-Tree algorithm used for unsorted data.
First created through urgency and adrenaline by Aaron Quinlan Spring 2009. Maintained by the Quinlan Laboratory at the University of Virginia.
- Lead developers: Aaron Quinlan, Hao Hoou, Brent Pedersen, Neil Kindlon
- Significant contributions: Assaf Gordon, Royden Clark, John Marshall, Brent Pedersen, Ryan Dale
- Repository: https://github.com/arq5x/bedtools2
- Stable releases: https://github.com/arq5x/bedtools2/releases
- Documentation: http://bedtools.readthedocs.org
- License: Released under MIT license
Please cite the following article if you use BEDTools in your research:
- Quinlan AR and Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 6, pp. 841–842.
Also, if you use pybedtools, please cite the following.
- Dale RK, Pedersen BS, and Quinlan AR. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics (2011). doi:10.1093/bioinformatics/btr539