This is PMET.
.
├── 00_binary_compile.sh
├── 01_homotypic_intervals.sh
├── 01_homotypic_promoters.sh
├── 01_homotypic_promoters_with_distance_to_tss.sh
├── 02_heterotypic_intervals.sh
├── 02_heterotypic_promoters.sh
├── 02_heterotypic_promoters_single_CPU.sh
├── 03_test_new_fimo.sh
├── 04_new_fimo_vs_old_fimo_plus_index.sh
├── 05_heatmap.R
├── LICENSE.md
├── data
├── results
├── scripts
├── src
│ ├── indexing # orginal pmet indexing with few changes
│ ├── indexing_c # pmet indexing in C, only for functionalites validation no real use
│ ├── meme-5.5.3 # new fimo with pmet indexing integrated
│ ├── pmet # orginal pmet with few changes
│ └── pmetParallel # pmet with parallel ability (defalut use for real analysis)
├── visualize_pmet_php
└── readme.md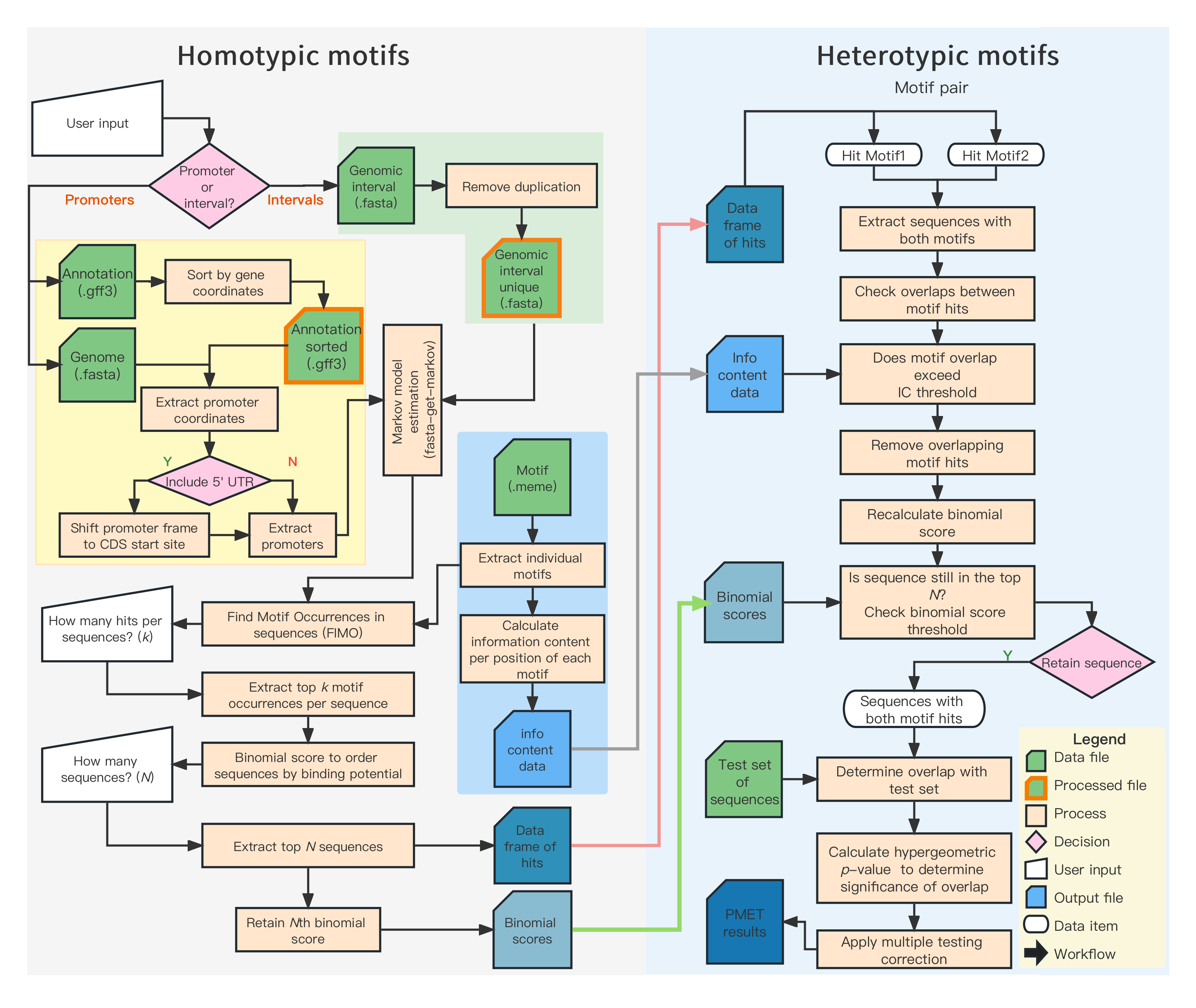
The purpose of this script is to
- assign execute permissions to all users for bash and perl files
- compile binaries needed by Shiny app
- install python package
chmod a+x 00_binary_compile.sh
bash 00_binary_compile.shBoth are writen in C++, source code can be found in src/indexing and src/pmetParallel.
If necessary, it is possible to compile PMET index and pmet in different OS.
Compile in one bash
chmod a+x 00_binary_compile.sh
bash 00_binary_compile.shAfter running the bash, all needed binary tools will be put in the scripts folder.
chmod a+x 01_homotypic_promoters.sh
chmod a+x 02_heterotypic_promoters.sh
bash 01_homotypic_promoters.sh
bash 02_heterotypic_promoters.shchmod a+x 01_homotypic_intervals.sh
chmod a+x 02_heterotypic_intervals.sh
bash 01_homotypic_intervals.sh
bash 02_heterotypic_intervals.shBefore running PMET index, we need to run FIMO to find all the homotypic motifs, and then PMET index will use the results from FIMO to run. This process will consume IO resources.
IO is expensive.
To mitigate the IO resource consumption associated with FIMO and the PMET index, we aim to integrate the capabilities of the PMET index directly into FIMO. Details of this integration can be found in the src/meme-5.3.3directory.
For instance, when querying 113 motif hits on the promoter of the Arabidopsis thaliana genome, the improved FIMO (referred to as NEW FIMO) can reduce write operations by 30GB and read operations by the same amount.
chmod a+x 03_test_new_fimo.sh
bash 03_test_new_fimo.shUsing the hardware specifications listed below, the traditional combination of FIMO and the PMET index takes more than double the time compared to using NEW FIMO. On a mechanical hard drive, this time difference can be amplified, possibly reaching 5 to 10 times.
- Single core processor
- Intel i9-12900K
- Samsung 980 Pro SSD
An additional consideration is the potential to divide the meme files (motifs) into segments and employ GNU Parallel for concurrent processing. This approach would decrease run times. Moreover, it would amplify the efficiency of NEW FIMO. Given that IO resources are finite, the IO resource usage of the combined FIMO and PMET index increases multiplicatively. No less time with more threads.
In contrast, NEW FIMO circumvents this issue entirely.
3.1 Install GNU Parallel
GNU Parallel helps PMET index (FIMO and PMET index) to run in parallel mode.
sudo apt-get install parallel
# Put GNU Parallel silent
parallel --citation3.2 Install The MEME Suite (FIMO and fasta-get-markov)
# cd a folder you want to put the software
wget https://meme-suite.org/meme/meme-software/5.5.2/meme-5.5.2.tar.gz
tar zxf meme-5.5.2.tar.gz
cd meme-5.5.2
./configure --prefix=$HOME/meme --enable-build-libxml2 --enable-build-libxslt
make
make test
make installAdd following into bash profile file.
# assuming you put meme folder under your home folder
export PATH=$HOME/meme/bin:$HOME/meme/libexec/meme-5.5.2:$PATH3.3 Install samtools
Install from conda or mamba:
conda install -c bioconda samtoolsInstall from source:
assuming you create a directory named
samtoolsin home directory (~) and install samtools there.
wget https://github.com/samtools/samtools/releases/download/1.17/samtools-1.17.tar.bz2
cd samtools-1.17 # and similarly for bcftools and htslib
./configure --prefix=$HOME/samtools
make
make install
# Add following into bash profile file or .zshrc (if zsh used).
# assuming you put samtools-1.17 folder under your home folder
export PATH=$HOME/samtools/bin:$PATH3. 4 Install bedtools
conda install -c bioconda bedtoolsor
# Debian/Ubuntu
apt-get install bedtools
# Fedora/Centos
yum install BEDToolsor
wget https://github.com/arq5x/bedtools2/releases/download/v2.29.1/bedtools-2.29.1.tar.gz
tar -zxvf bedtools-2.29.1.tar.gz
cd bedtools2
makepip install numpy
pip install pandas
pip install scipy
pip install bio
pip install biopython