SmartSeq2 scRNASeq QC Metrics
Sequencing QC metrics and its visualization can not only provide overall view of quality for a experiment but also play important role in troubleshooting,experiment improvement. In this section, we will have a overall view discussion of several QC metrics which were generated by scRNASeq pipeline
Tables of metrics can provide an overview of alignment statistics,rna sequencing quality and more.
Alignment metrics can be used to provide overall idea of the quality of alignment for your libraries. One of important metrics is PCT_PF_ALIGNED which calculates the percentage of reads mapped to reference genome. Another important metrics is PF_MISMATCH_RATE, which can provide overall alignment quality. Example shown below.

RNA metrics provide important summary based on gene annotation. PCT_USABLEBASES measures the percentage of bases mapped to transcriptome(mRNA+UTR regions). This metrics provide overall view of quality of RNA sequencing. Higher values in PCT_INTRONIC_BASES, PCT_/INERGENIC_BASES and PCT_RIBOSOMAL_BASES indicate lower quality or degraded in RNA. High in MEDIAN_3PRIME_BIAS(>1) also indicates high chance to have degraded RNA. Example shown below.

This metrics provides basic information on insert sizes for paired-end library. This metrics can be used to ensure that pair-end libraries are constructed as expected. Example shown below.

This metrics provides level of duplication(post alignment). It is coordinates based method, not raw sequencing(fastq) data based method. Example shown below.

In this task, we applied a scRNA-Seq pipeline on a published dataset GSE47872. We collected metrics of all single cell samples, include primary Glioblastoma cell and Gliomasphere Cell Line cells. Sample counts are listed below:
| 25bp | 100bp | |
|---|---|---|
| Glioblastoma | 581 | 96 |
| Gliomasphere Cell Line | 195 | 0 |
We collected all metrics together and generated one table. We visualized several important metrics shown as below.
First,We examined difference in metrics between primary cell and cell line and we only used metrics of 25bp length samples.
TOTAL_READS metrics' density plot shown in figure. Primary cell samples show slightly narrow distribution of reads counts than cell line samples.

PCT_PF_READS_ALIGNED density plot. Overall, both types of samples yield ~75% alignment rate and both have unusual peaks at 25% alignment rate.

PF_MISMATCH_RATE density plot. Primary cell sample's PF_MISMATCH_RATE have two peaks, one overlaps with cell line samples and another one locates at the lower end.

PCT_USABLE_BASES density plot.

PCT_RIBOSOMAL_BASES density plot. Both types of samples yield good percentage of ribosomal bases.

MEDIAN_CV_COVERAGE density plot. The median coefficient of variation (CV) or stdev/mean for coverage values of the 1000 most highly expressed transcripts. Low values is ideal. Primary cell samples have wider distribution in CV which can also means a wider distribution around mean coverage.

MEDIAN_3PRIME_BIAS and MEDIAN_5PRIME_BIAS density plots. Both type of samples show low bias at 5' and 3' end but there is a long tail extended to high bias region which indicates there is chance of degradation in some of cells.


MEDIAN_INSERT_SIZE and MEDIAN_ABSOLUTE_DEVIATION density plots. both types fall into expected region, 200~400bp


PERCENT_DUPLICATION density plot. Both types show on average ~10% of duplication rate and a subset of cells show high duplication rate ~75%.

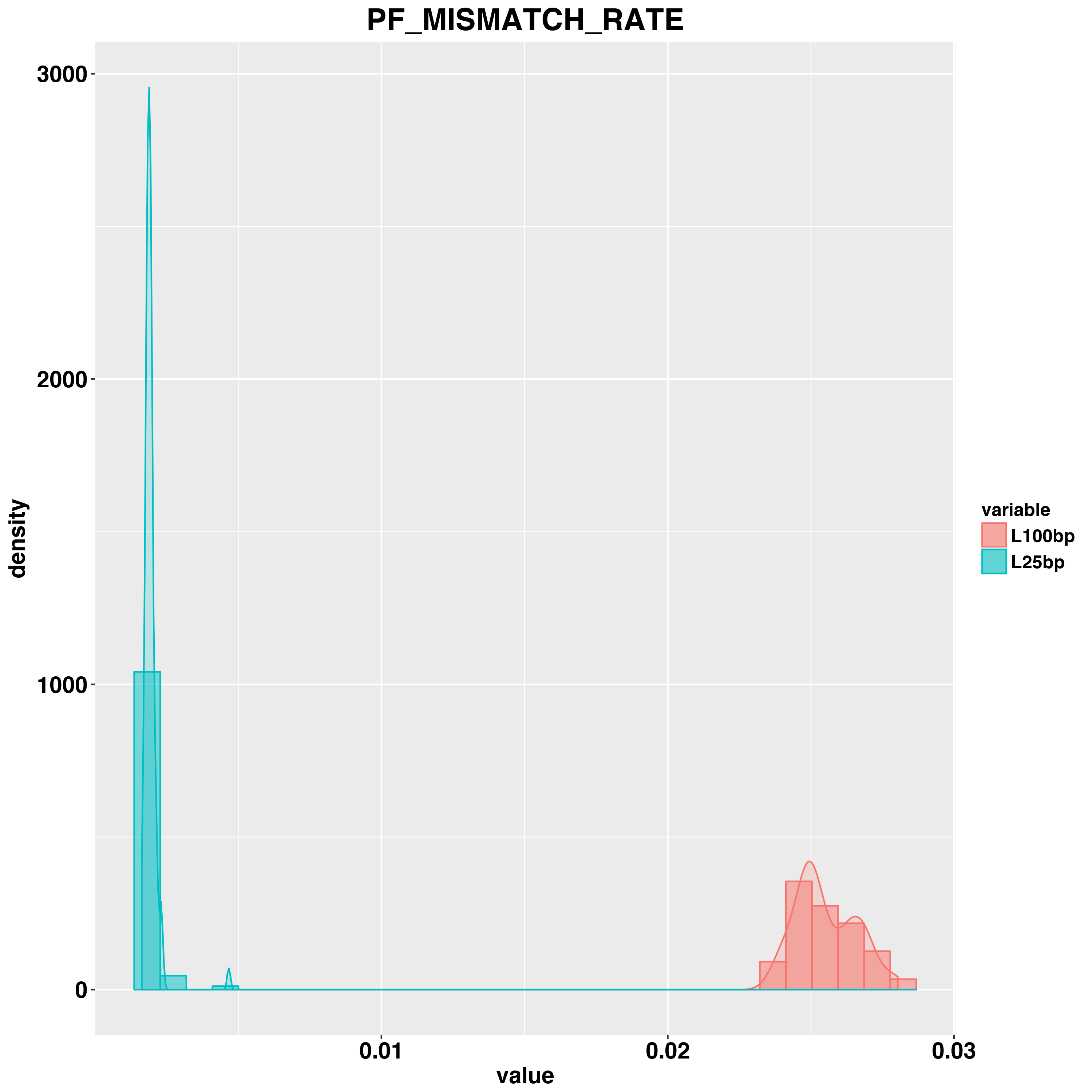
Then we examined 96 paired samples and samples in a pair were sequenced from the same library but yield at different read length, 25bp(L25bp) and 100bp(L100bp).
For alignment, we examined PCT_PF_READS_ALIGNED, PF_MISMATCH_RATE and MEDIAN_CV_COVERAGE between paired L100bp and L25bp samples. L100bp and L25bp have similar PCT_PF_READS_ALIGNED. L100bp samples have lower value of CV (which is ideal). L100bp samples have much higher PF_MISMATCH_RATE compared to L25bp. The cause for this difference is unclear, maybe due to the parameters we used to run STAR alignment.

For RNA metrics, we examined PCT_USABLE_BASES and PCT_INTRONIC_BASES, PCT_RIBOSOMAL_BASES. Since paired L100bp and L25bp samples are actually sequenced from the same library, it is not surprising to observe the high consistency in rna metrics.



For Insertion Size metrics, we examined MEDIAN_INSERT_SIZE. This metrics is high consistent between L100bp and L25bp samples.

For Duplication metrics, we examined PERCENT_DUPLICATION. L100bp show lower duplication rate.
