A repository of update in molecular dynamics field by recent progress in machine learning and deep learning. Those efforts are cast into the following categories:
- Learn force field or molecular interactions
- Enhanced sampling methods
- Learn collective variable
- Learn kinetic model
- Capture dynamics of molecular system
- Map between all atoms and coarse grain
- Design proteins
- Protein-ligand prediction for drug discovery
- Modeling Reactive Potential Energy Surface
Molecular Graph Convolutions: Moving Beyond Fingerprints
Steven Kearnes, Kevin McCloskey, Marc Berndl, Vijay Pande, Patrick Riley. (2016)
This paper from Standford Univ and Google proposed graph representation of molecules and graph convolution to capture the interactions in the molecule. The authors used a weave module, where the atom feature and edge feature are weaved to preserve invariance of atom and pair permutation. They used Gaussian membership functions to preserve overall order invariance.
An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2
NongnuchArtrith, Alexander Urban. (2016)
The authors from UC Berkeley developed open-source atomic energy network package, based on Behler-Parrinello machine learning potential, which uses multilayer perceptron to learn the potential of molecules. The atomic coordinates are transformed into invariant representation of the local atomic environments and potential is trained on such representation. The authors applied the model to TiO2, ZrO2, and alpha-PbO2.
Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models
Garrett B. Goh, Charles Siegel, Abhinav Vishnu, Nathan O. Hodas, Nathan Baker. (2017)
The authors from Pacific Northwest National Laboratory developed this computer vision-based model for chemicals. By converting SMILES strings to images and encoding atom properties through color channels, the model slightly outperforms ECFP fingerprints-based deep NN in activity and solvation, and slightly underforms in toxicity prediction.
Machine learning prediction errors better than DFT accuracy
Felix A. Faber, Luke Hutchison, Bing Huang, Justin Gilmer, Samuel S. Schoenholz, George E. Dahl, Oriol Vinyals, Steven Kearnes, Patrick F. Riley, O. Anatole von Lilienfeld. (2017)
The authors from Univ of Basel and Google used elastic network, bayesian regression, random forest, kernel ridge regression, gated graph NN, graph convolutions to predict QM9 data set. The representations are Coulomb matrix, BAML (bonds, angles, machine learning), ECFP4 (extended connectivity fingerprints), MARAD (molecular atomic radial angular distribution), HD, HDA, HDAD (histogram methods). They demonstrated the machine learning methods have smaller error than DFT error.
Quantum-Chemical Insights from Deep Tensor Neural Networks
Kristof T. Schütt, Farhad Arbabzadah, Stefan Chmiela, Klaus R. Müller, Alexandre Tkatchenko. (2017)
The authors from Technische Universitat Berlin, Korea Univ, Fritz-Haber-Institut der Max-Planck-Gesellschaft and Univ of Luxembourg developed DTNN. The network used atom features and edge features for input. Edges are processed by Gaussian expansion. The edges and atoms interact through an interaction module through tensor multiplications. The authors applied this to predict chemical potentials, ring stability of molecules etc.
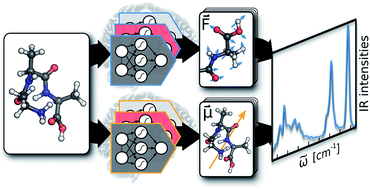
Machine learning molecular dynamics for the simulation of infrared spectra
Michael Gastegger, Jörg Behler, Philipp Marquet. (2017)
The authors from Univ. of Vienna and Universität Göttingen developed a molecular dipole moment model based on environment-dependent NN and combined with NN potential approach of Behler and Parrinello for ab inito MD. As an application, they obtained accurate models for predicting infrared spectra.
ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost
J. S. Smith, Isayev, A. E. Roitberg. (2017)
This paper from Univ. of Florida and Univ. of North Carolina presented ANI-1, which used Behler and Parrinello symmetry functions to build single-atom atomic environment vectors (AEV) as molecular representation. This is similar to the context representation of work in NLP.
ElemNet: Deep Learning the Chemistry of Materials From Only Elemental Composition
Dipendra Jha, Logan Ward, Arindam Paul, Wei-keng Liao, Alok Choudhary, Chris Wolverton & Ankit Agrawal. (2018)
The authors from Northwestern Univ, Univ of Chicago developed ElemNet, which takes elemental compositions and used 17-layer MLP architecture to predict DFT-computed formation enthalpies for quantuam materials. The authors visualized 1st, 2nd, and 8th layers of the network to elucidate the chemistry insights that the model learned.
Towards exact molecular dynamics simulations with machine-learned force fields
Stefan Chmiela, Huziel E. Sauceda, Klaus-Robert Müller, Alexandre Tkatchenko. (2018)
The authors from Technische Universität Berlin, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Korea Univ, and Univ of Luxembourg developed a kernel-based symmetric gradient-domain ML (sGDML) model to reproduce global force fields at CCSD(T) level of accuracy. It allows converged MD simulations with fully quantized electrons and nuclei. This work built on their previous work - GDML, with symmetry imposed in the current sGDML. The authors constructed FF in this 2019 JCP paper.
Applying machine learning techniques to predict the propertiesof energetic materials
Daniel C. Elton, Zois Boukouvalas, Mark S. Butrico, Mark D. Fuge, Peter W. Chung. (2018)
The authors from Univ of Maryland applied several machine learning methods (KRR, ridge, SVR, RF, k-nearest neighbor) based on features (sum over bonds, custom descriptors, Coulomb matrices, Bag of Bonds, and fingerprints). They concluded the best featurization is sum over bonds and best model is kernel ridge regression.
Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics
Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, Weinan E. (2018)
The authors from Peking Univ., Princeton Univ., and Institute of Applied Physics and Computational Mathematics, China developed DeepMD method based on a many-body potential and interatomic forces generated by NN, which is trained with ab initio data.
Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials
Peter Bjørn Jørgensen, Karsten Wedel Jacobsen, Mikkel N. Schmidt. (2018)
This paper from Univ. of Denmark extended neural message passing model with an edge update NN, so that information exchanges between atoms depend on hidden state of the receiving atom. They also explored ways to construct the graph.
SchNet – A deep learning architecture for molecules and materials
K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko, K.-R. Müller. (2018)
This paper from Technische Universita ̈t Berlin, Univ. of Luxembourg, Max Planck Institute, and Korea University presented SchNet, a variant of DTNN to learn the molecular properties and studied local chemical potential and the dynamics of C20-fullerene.
Pixel Chem: A Representation for Predicting Material Properties with Neural Network
Shuqian Ye, Yanheng Xu, Jiechun Liang, Hao Xu, Shuhong Cai, Shixin Liu, Xi Zhu.(2019)
The authors designed a Pixel Chemistry network to learn a representation for predicting molecular properties. The authors proposed three new matrices, which reflect charge transfer ability, bond binding strength, and Euclidean distances between atoms. They also designed an angular interaction matrix A, describes the interaction between two atomic orbitals.
Message-passing neural networks for high-throughput polymer screening
Peter C. St. John1, Caleb Phillips, Travis W. Kemper, A. Nolan Wilson, Yanfei Guan, Michael F. Crowley, Mark R. Nimlos, Ross E. Larsen. (2019)
This paper from National Renewable Energy Lab, USA, used message-passing NN to predict polymer properties for screening purpose. They focused on larger molecules and tested the model with/without 3D conformation information, since accurate 3D structure calculation is also expensive.
Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network
Roman Zubatyuk, Justin S. Smith, Jerzy Leszczynski and Olexandr Isayev. (2019)
This paper from Univ. of North Carolina, Los Alamos National Lab, and Jackson State Univ presented AIMNet to leearn implicit solvation energy in MNSol database. Atoms in molecules are embedded and interact with each in several layers.
LanczosNet: Multi-Scale Deep Graph Convolutional Networks
Renjie Liao, Zhizhen Zhao, Raquel Urtasun, Richard S. Zemel. (2019)
The authors from Univ. of Toronto, Uber ATG, Vector Institute, UIUC and Canadian Institute of Advanced Research developed this spectral-based graph NN, which uses Lanczos algorithms to construct low rank approximations of the graph Laplacian. They benchmarked the model on citation networks and QM8 dataset.
Molecule-Augmented Attention Transformer
Łukasz Maziarka, Tomasz Danel, Sławomir Mucha, Krzysztof Rataj, Jacek Tabor, Stanisław Jastrzebski. (2019)
The authors from Jagiellonian Univ, Ardigen and New York Univ designed this MAT graph NN model with self-attention mimicking the Transformer, consisting of multiple blocks of layer norm, multi-head self-attention, and residual net. The model achieved comparable or better results on BBBP and FreeSolv datasets comparing with MPNN.
Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules
Benjamin Sanchez-Lengeling, Jennifer N. Wei, Brian K. Lee, Richard C. Gerkin, Alán Aspuru-Guzik, Alexander B. Wiltschko. (2019)
This paper from Google, Arizona State Univ, Univ of Toronto, Vector Institute, Canadian Institute for Advanced Research used MPNN (message passing NN) based on graph representation, to predict quantitative structure-odor relationship (QSOR), very similar to QSAR. The model out-performed molecular fingerprint-based methods. The authors showed their learned embeddings from GNN capture a meaningful odor space representation.
ProDyn0: Inferring calponin homology domain stretching behavior using graph neural networks
Ali Madani, Cyna Shirazinejad, Jia Rui Ong, Hengameh Shams, Mohammad Mofrad. (2019)
This paper from UC Berkeley used MPNN and residual gated graph convnets to predict the pattern and mode of SMD (steered MD) simulation results. The authors created this data set of 2020 mutants of calponin homology domain (CH, an actin-binding domain) with SMD simulation results. Capturing the force between CH domains is capturing molecular interactions between amino acid residues.
Reinforced dynamics for enhanced sampling in large atomic and molecular systems
Linfeng Zhang, Han Wang, Weinan E. (2018)
This paper from Peking Univ., Princeton Univ, and IAPCM, China used used a vanilla NN to predict the mean force on CV as a construction of FES, which is then used as a biasing potential in metadynamics on the CV. The uncertainty of the FES is used to determine the whether to use those parts for training NN or not. The authors benchmarked on a full-atom explicit solvent system of Ala dipeptide and tripeptide, and a Ala 10-mer peptide.
Reinforcement Learning Based Adaptive Sampling: REAPing Rewards by Exploring Protein Conformational Landscapes
Zahra Shamsi, Kevin J. Cheng, Diwakar Shukla. (2018)
This paper from UIUC used reinforcement learning to adaptively biase the sampling potential. The action in this RL problem is to pick new structures to start a swarm of simulations, and the reward function is how far order parameters sample the landscape.
Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning
Frank Noé, Simon Olsson, Jonas Köhler, Hao Wu. (2019)
This paper from Freie Universität Berlin, Rice Univ and Tongji Univ used a generative model, Boltzmann generator machine, to generate unbiased equilibrium samples from different metastable states in one shot. This model is said to overcome rare event-sampling problems in many-body systems.
Targeted Adversarial Learning Optimized Sampling
Justin Zhang, Yi Isaac Yang, Frank Noé (2019)
The authors from Freie Universität Berlin use adversarial training to steer a molecular dynamics ensemble towards a desired target distribution, overcoming rare-event sampling problems.
Neural networks-based variationally enhanced sampling
Luigi Bonati, Yue-Yu Zhang, Michele Parrinello. (2019)
The authors from ETH Zurich, Universita della Svizzera italiana, MARVEL (Switzerland), and Italian Institute of Technology presented a NN-based bias potential for enhanced sampling, building on their previous work of variationally enhanced sampling. Deep learning provides an expressive tool for mapping from CV to actual bias potential.
DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding
Hyungro Lee, Heng Ma, Matteo Turilli, Debsindhu Bhowmik, Shantenu Jha, Arvind Ramanathan. (2019)
The authors from Rutgers Univ, Argonne, Oak Ridge, Brookhaven National Lab developed DeepDriveMD, which uses MD simulation to generate molecular samples, use convoluted VAE based on those samples to learn the latent space, cluster the latent space and find rare regions to continue spawn new simulations. Another highlight of the work is their ensemble execution of the model on Summit.
Machine Learning Based Dimensionality Reduction Facilitates Ligand Diffusion Paths Assessment: A Case of Cytochrome P450cam
Jakub Rydzewski, and Wieslaw Nowak. (2016)
The authors from Nicolaus Copernicus University showed how t-distributed stochastic neighbor embedding (t-SNE) can be applied to analyze the process of camphor unbinding from cytochrome P450cam via multiple reaction pathways.
Transferable Neural Networks for Enhanced Sampling of Protein Dynamics
Mohammad M. Sultan, Hannah K. Wayment-Steele, Vijay S. Pande. (2018)
The authors from Stanford Univ used variational autoencoder with time-lagged information to learn the collective variable in latent space. They then used the latent space representation in well-tempered ensemble metadynamics. The authors showed such learned latend space is transferrable for proteins with certain mutations or between force fields.
Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration
Wei Chen, Andrew L. Ferguson. (2018)
The authors from UIUC did on-the-fly CV discovery by using an autoencoder, so-called "chicken-and-egg" problem. The bottleneck in autoencoder maps the "intrinsic manifold". Each time after discovering the CV, the model did boundary detection and then did umbrella sampling to further explore the configurational space. They dealt with translational invariance by removing center of mass movement and dealt with rotational invariance by data augmentation. The model was benchmarked on alanine dipeptide and Trp-cage.
Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics
Christoph Wehmeyer, Frank Noé. (2018)
The authors from Freie Universität Berlin built time-lagged autoencoders to learn the slow collective variables. They show that time-lagged autoencoders are a nonlinear generalization of the time-lagged independent component analysis (TICA) method.
Reweighted autoencoded variational Bayes for enhanced sampling (RAVE)
João Marcelo Lamim Ribeiro, Pablo Bravo, Yihang Wang, and Pratyush Tiwary. (2018)
This paper from Univ of Maryland and Pontificia Universidad Catolica de Chile used variational autoencoder and Bayes theorem to find the reaction coordinates and approapriate weights. Kullback-Leibler divergence is calculated between this latent space distribution and the distribution of various trial reaction coordinates sampled from the simulation.
Learning protein conformational space by enforcing physics with convolutions and latent interpolations
Venkata K. Ramaswamy, Chris G. Willcocks, Matteo T. Degiacomi. (2019)
This paper from Durhan Univ designed a CNN-based autoencoder to learn a continuous latent space for protein conformations. Based on the latent space, they derived a transition path between two states. The authors also augmented the network with MD simulation data, incorporating physics-based constraints, achieving high accuracy.
Enhancing Biomolecular Sampling with Reinforcement Learning: A Tree Search Molecular Dynamics Simulation Method
Kento Shin, Duy Phuoc Tran, Kazuhiro Takemura, Akio Kitao, Kei Terayama, Koji Tsuda. (2018)
The authors from Univ of Tokyo, Tokyo Institute of Tech, RIKEN, Kyoto Univ, National Institute for Material Science developed tree search MD (TS-MD). To sample the transition pathway from a given initial configuration to a target configuration, the authors performed short MD simulations with new random velocities and considered snapshots as nodes in the tree. They used upper confidence bounds for trees (UCT) to solve the exploration-exploitation dilemma.
Nonlinear discovery of slow molecular modes using state-free reversible VAMPnets
Wei Chen, Hythem Sidky, Andrew L. Ferguson. (2019)
The authors from UIUC and Univ of Chicago introduced SRV, state-free reversible VAMPnets to learn nonlinear CV approximants. The work built on VAMPNet (variational approach for Markov processes networks). SRV learns the first few slow eigenfunctions of the spectral decomposition of the transfer operator, which evolves probability distribution at equilibrium through time.
Past–future information bottleneck for sampling molecular reaction coordinate simultaneously with thermodynamics and kinetics
Yihang Wang, João Marcelo Lamim Ribeiro, Pratyush Tiwary. (2019)
The authors from Univ of Maryland used variational inference implemented in deep neural networks to infer reaction coordinates/CV. To sample the rare event, the authors took analogue of predictive information bottleneck, trying to maximize the prediction of future by utilizing the informaiton from the past.
Artificial Intelligence Assists Discovery of Reaction Coordinates and Mechanisms from Molecular Dynamics Simulations
Hendrik Jung, Roberto Covino, Gerhard Hummer. (2019)
The authors from Max Planck Institute of Biophysics and Goethe Univ introduced an NN-based model to find the reaction coordinates. Based on the transition path sampling (TPS) theory, the authors did MD simulations, built transition path ensemble, find reaction coordinates and do more MD simulations.
VAMPnets for deep learning of molecular kinetics
Andreas Mardt, Luca Pasquali, Hao Wu, Frank Noé (2018)
The authors from Freie Universität Berlin employ the variational approach for Markov processes (VAMP) to develop a deep learning framework for molecular kinetics using neural networks, dubbed VAMPnets. A VAMPnet encodes the entire mapping from molecular coordinates to a Markov state model (MSM), thus combining the MSM whole data processing pipeline in a single end-to-end framework.
Neural Mode Jump Monte Carlo
Luigi Sbailò, Manuel Dibak, Frank Noé (2019)
The authors from Freie Univ Berlin and Rice Univ developed this NN-based Monte Carlo jump scheme. To sample both locally and globally metastable states, a local proposal scheme and a neural proposal scheme are applied respectively. The neural proposals connect different metastable states, where the end point is sampled from a predefined probability distribution. The neural network is trained in an unsupervised fashine, as training set gradually grows.
Equivariant Hamiltonian Flows
Danilo Jimenez Rezende, Sébastien Racanière, Irina Higgins, Peter Toth. (2019)
This paper from Google uses Lie algebra to prove what hamiltonian flow learns and how addition of symmetry invariance constraints can improve data efficiency.
Equivariant Flows: sampling configurations formulti-body systems with symmetric energies
Jonas Köhler, Leon Klein, Frank Noé. (2019)
This paper from Freie Universität Berlin model flows that have symmetries in the energy built in, such as roto-translational and permutational invariances, as a system of interacting particles. Can be used both for learning particle dynamics and sampling equilibrium states.
Symplectic ODE-NET: learning Hamiltonian dynamics with control
Yaofeng Desmond Zhong, Biswadip Dey, Amit Chakraborty. (2019)
This paper from Princeton University and Siemens Corp infers the dynamics of a physical system from observed state trajectories. They embedded high dimensional coordinates into low dimensions and velocity into general momentum.
Hamiltonian Neural Networks
Sam Greydanus, Misko Dzamba, Jason Yosinski. (2019)
This paper from Google, PetCube and Uber trains models to learn conservation law of Hamiltonian in unsupervised way.
Symplectic Recurrent Neural Networks
Zhengdao Chen, Jianyu Zhang, Martin Arjovsky, Léon Bottou. (2019)
The authors from NYU, Tianjin University, and Facebook proposes SRNN to capture the dynamics of physical systems from observed trajectories.
Physical Symmetries Embedded in Neural Networks
M. Mattheakis, P. Protopapas, D. Sondak, M. Di Giovanni, E. Kaxiras. (2019)
The authors from Harvard and Polytechnic Milan used symplectic neural network to embed physics symmetry in the neural network to characterize the dynamics.
Neural Canonical Transformation with Symplectic Flows
Shuo-Hui Li, Chen-Xiao Dong, Linfeng Zhang, Lei Wang. (2019)
The authors from CAS, Princeton Univ., and Songshan Lake Materials Lab constructed canonical transformation with symplectic neural networks. Such formulations help understand the physical meaning of latend space in the model. The authors applied this to learn slow CV of analine dipeptide and conceptual compression of MNIST dataset.
Stochastic normalizing flows
Hao Wu, Jonas Köhler, Frank Noé. (2020)
The authors from Tongji Univ, Freie Universita ̈t Berlin, and Rice Univ added stochasticity into normalizing flow, by MCMC or Langevin dynamics. Normalizing flow (NF) is an invertible mapping between two distributions and well-known for its expressive power. Stochasticity improves expressiveness of NF. The authors benchmarked on double-well potential, alanine dipeptide, variational inference.
Targeted free energy estimation via learned mappings
Peter Wirnsberger, Andrew J. Ballard, George Papamakarios, Stuart Abercrombie, Sébastien Racanière, Alexander Pritzel, Danilo Jimenez Rezende, Charles Blundell. (2020)
The authors from DeepMind used normalizing flow for mapping in targeted free energy perturbation (TFEP). TFEP was developed by Jarzynski in 2002 using an invertible mapping defined on configuration space transporting distribution A to distribution A', so that A' can be close to B. Such a mapping is now done by ML generative model, normalizing flow. The authors benchmarked on a box of solvent particles to calculate free energy of growing the solute radius from Ra to Rb.
Differentiable Molecular Simulations for Control and Learning
Wujie Wang, Simon Axelrod, and Rafael Gómez-Bombarelli. (2020)
The authors from MIT and Harvard demonstrate the use of automatic differentiation to devise simulation protocols, learn from macroscopic distribution function, and design control protocols for molecular quantum dynamics.
Machine Learning of coarse-grained Molecular Dynamics Force Fields
Jiang Wang, Simon Olsson, Christoph Wehmeyer, Adrià Pérez, Nicholas E. Charron, Gianni de Fabritiis, Frank Noé, Cecilia Clementi. (2018)
The authors from Rice University, Freie Universität Berlin, and Universitat Pompeu Fabra presented CGnet which learns coarse grain force field by using variational force matching. They also recast force-matching as a machine learning problem, allowing to decompose the force matching error into bias, variance and noise. They demonstrated the model performance on dialanine peptide simulation and Chignolin folding/unfolding in water.
DeePCG: Constructing coarse-grained models via deep neural networks
Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, and Weinan E. (2018)
The authors from Peking Univ, Princeton Univ, and IAPCM, China presented DeepCG to construct a many-body CG potential. The authors applied this to liquid water and did CG simulation starting from an atomistic simulation at ab inito level.
Adversarial-Residual-Coarse-Graining: Applying machine learning theory to systematic molecular coarse-graining
Aleksander E. P. Durumeric, Gregory A. Voth. (2019)
The authors from Univ. of Chicago employed generative adversial network (GAN) for systematic molecular coarse-graining. They showed that the resulting framework can rigorously parameterize CG models containing CG sites with no prescribed connection to the reference atomistic system.
Coarse-graining auto-encoders for molecular dynamics
Wujie Wang and Rafael Gómez-Bombarelli. (2019)
The authors from MIT propose an auto-neoder based method to parameterize coarse-grained variables from data using discrete variable reparametrization. They also demonstrate the use of Graph Neural Networks to fit Coarse-Grained models by force matching.
(Though this part is less connected to MD simulation, some of the ML-based protein design algorithms are actually inditectly learning the potential energy of proteins. So we keep a small portion here.)
Learned Protein Embeddings for Machine Learning
Kevin K. Yang, Zachary Wu, Claire N. Bedbrook, Frances H. Arnold. (2018)
The authors from Caltech used doc2vec k-mers method in NLP to pretrain the embedding and further used task-specific supervised learning to learn the embedded vector for amino acids. The training datasets have sizes ranging from 81 ~ 261, regarding plasma membrane localization, thermostability, rhodopsin peak absorption wavelength etc. The authors showed the embedding outperforms one-hot encoding, mismatch kernel, feature-engineering method like ProFET and AAIndex.
Generative Models for Graph-Based Protein Design
John Ingraham, Vikas K. Garg, Regina Barzilay, Tommi Jaakkola. (2019)
This paper from MIT used generative graph model to design proteins. View this as a reverse problem of protein folding/structure prediction, the authors showed their approach efficiently captures the long-range interactions that are distant in sequence but local in 3D structure.
Learning Protein Sequnce Embedding Using Information from Structure
Tristan Bepler, Bonnie Berger. (2019)
The authors from MIT developed this ELMo-like sequence embedding method by incorporating language model and structural inference. Language model predict the next amino acid in each direction, thus it captures the amino acid local context. The learned hidden states are then combined with input sequences and go through 3 layers of LSTMs for soft symmetric alignment and structural similarity prediction. Since the protein language is more at 3D structural level, the structural similarity prediction is a good task to supervise embedding training.
AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
Izhar Wallach, Michael Dzamba, Abraham Heifets. (2015)
The authors from Atomwise Inc developed AtomNet using 3D CNN to predict bioactivity (effectiveness of ligands). The input features are 3D grids of the protein-ligand complex, with center of mass centered at Cartesian origin. Four layers of convolution layers were applied, followed with two fully-connected layers. The model was benchmarked on DUDE and ChEMBL-20 PMD datasets.
Boosting Docking-Based Virtual Screening with Deep Learning
Janaina Cruz Pereira, Ernesto Raúl Caffarena, Cicero Nogueira dos Santos. (2016)
The authors from Fiocruz and IBM Watson developed DeepVS, a DL-based docking virtural screening. Utilizing inputs of protein-ligand complex, the authors used embeddd vectors to represent atom neighbors (type, charge, distance) and amino acid neighbors. The inputs then pass through convolution layers to extract the important features and finally predict the docking ranking list.
Protein–Ligand Scoring with Convolutional Neural Networks
Matthew Ragoza, Joshua Hochuli, Elisa Idrobo, Jocelyn Sunseri, David Ryan Koes. (2017)
The authors from Univ of Pittsburgh and College of New Jersy developed a 3D CNN model to predict protein-ligand binding score. The authors treated each atom type as a color channel and gridize the 3D space to convert to a computer vision problem. Atom type information is represented as a density function around atom center. Five convolution layers were used, followed by fully connected layers. The authors benchmarked on CSAR and DUDE datasets.
Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity
Joseph Gomes, Bharath Ramsundar, Evan N. Feinberg, Vijay S. Pande. (2017)
The authors from Stanford Univ developed ACNN (atomistic CNN) for predicting protein-ligand affinity. To represent the local chemical environment, they used atome type convolution to integrate local environment, and radial pooling on distance matrix with neighbor list construction, similar to the graph representation. The output of model is energy, the authors also integrate thermodynamic cycle to learn the binding free energy from G_{lig}, G_{prot}, and G_{complex}. The implementation was compared with grid featurizer, GCNN and ECFP fingerprint methods on PDBbind dataset.
Molecular de-novo Design through Deep Reinforcement Learning
Marcus Olivecrona, Thomas Blaschke, Ola Engkvist and Hongming Chen. (2017)
The authors from AstraZeneca developed a RNN-based reinforcement learning model to do de-novo drug design for desired properties. The input are SMILE strings with representation learned by RNN through predicting next character in the string. This pretrained RNN is then refined with policy-based RL to get more reward so that the properties of molecules are desired.
druGAN: An Advanced Generative Adversarial Autoencoder Model for de novo Generation of New Molecules with Desired Molecular Properties in silico
Artur Kadurin, Sergey Nikolenko, Kuzma Khrabrov, Alex Aliper, and Alex Zhavoronkov. (2017)
The authors from JHU, National Research Univ, Steklov Math Inst, Mail.Ru Group, Biogerontology Res Foundation, Moscow Inst of Physics and Tech, Kazan Federal Univ developed a GAN model, druGAN. The adversarial autoencoder (AAE) has molecular fingerprints as input and reconstruction output. Discriminator serves as teacher for the encoder to regularize the model. They also compared performance of AAE vs VAE.
Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks
Marwin H. S. Segler, Thierry Kogej, Christian Tyrchan, Mark P. Waller. (2017)
The authors from Westfal̈ische Wilhelms-Universitaẗ Münster, AstraZeneca and Shanghai Univ trained an RNN model based on SMILE strings to learn to generate molecules for a specific target. To learn the language model of drug molecules, they first trained it on a bigger dataset. Later they trained the model on smaller dataset to finetune it.
Mol2vec: unsupervised machine learning approach with chemical intuition
Sabrina Jaeger, Simone Fulle, Samo Turk. (2017)
The authors from BioMed X Innovation Center, Heidelberg developed Mol2Vec model to learn vector embedding for chemicals. They treated a molecule as a sentence and ECFP finger prints as words, utilizing CBOW and Skip-gram in Word2Vec as prediction tasks to learn the word embedding. This Mol2Vec was later combined with Prot2Vec to predict drug response.
Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening
Zixuan Cang,Lin Mu,Guo-Wei Wei. (2018)
The authors from Michigan State Univ used algebraic topology for ligand representation. Using persistent homology, the authors were able to extract molecular features at different levels. They also used different ways to represent the interactions inside the molecule (Rips or Alpha complex). This has close relation to the graph-based neural net works. Know more about persistent homology, this video may be useful.
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
Rafael Gómez-Bombarelli, Jennifer N. Wei, David Duvenaud, José Miguel Hernández-Lobato, Benjamín Sánchez-Lengeling, Dennis Sheberla, Jorge Aguilera-Iparraguirre, Timothy D. Hirzel, Ryan P. Adams, Alán Aspuru-Guzik. (2018)
The authors from Kyulux North America Inc, Harvard, UToronto, UCambridge, Google, Princeton Univ, and CIFAR designed a VAE(variational autoencoder)-style model to learn a continuous latent space for chemicals. The input is SMILE string, going through RNN or CNN to map to latent space, and later is reconstructed to SMILE strings. They also built a prediction model using latent space vectors to predict drug properties.
KDEEP: Protein−Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks
JoséJimeńez, MihaŠkalic, GerardMartínez-Rosell, and Gianni De Fabritiis. (2018)
The authors from Univ Pompeu Fabra and ICREA, Spain developed K_DEEP model to predict protein-ligand binding affinity by 3D convolution. The authors voxelized the protein-ligand complex and used a set of 3D descriptors, e.g., ionizable, hydrophobic, aromatic, H-bond acc/rec, etc. as the input features. They adapted SqueezeNet archietecture as the 3D CNN model. Rotational invariance is achieved by data augmentation rotating the input subgrids. The model was benchmarked on PDBbind and CSAR.
Development of a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes
Gabriela Bitencourt-Ferreira, Walter Filgueira de Azevedo. (2018)
The authors from PUCRS, Brazil combined Autodock scoring functions to predict the Gibbs free energy. They used methods like LASSO, Ridge, elastic net etc. to determine the weights for those polynomial functions. The authors also offered a dissection of the energy/scoring function terms in several software, e.g., Autodock4, Vina, MVD.
WideDTA: prediction of drug-target binding affinity
Hakime Öztürk, Elif Ozkirimli, Arzucan Özgür. (2019)
The authors from Bogazici Univ in Turkey developed an NLP-based method, WideDTA, for predicting ligand binding affinity. The 'words' of protein are 3-residue representations, while 'word' for ligand is 8-char subsequences of sliding window based on SMILE strings. Using CNN, the authors showed their model outperformed DeepDTA on KIBA dataset.
Learning from the Ligand: Using Ligand-Based Features to Improve Binding Affinity Prediction
Fergus Boyles, Charlotte M. Deane, and Garrett M. Morris. (2019)
The authors from UOxford developed a binding affinity prediction model including both protein-ligand structure and ligand-based property features. The former includes RF-Score and NNScore, while the latter includes 185 RDKit descriptors, eg., logP, molar refractivity etc. They showed those ligand-based features enhanced the performance.
ReSimNet: drug response similarity prediction using Siamese neural networks
Minji Jeon, Donghyeon Park, Jinhyuk Lee, Hwisang Jeon, Miyoung Ko, Sunkyu Kim, Yonghwa Choi, Aik-Choon Tan, Jaewoo Kang. (2019)
The authors from Korea Univ and Univ of Colorado developed ReSimNet to predict drug response similarity. Using a data-driven approach, the model was trained on drug-gene transcription profile dataset to predict CMap score of two compounds. It was later tested on ZINC15 dataset to show it is able to identify similar chemical compounds of a prototype drug.
Target-Specific Prediction of Ligand Affinity with Structure-Based Interaction Fingerprints
Florian Leidner, Nese Kurt Yilmaz, and Celia A. Schiffer. (2019)
The authors from Univ of Massachusette used gradient boosting method to predict HIV-1 protease inhibitor affinity. They used hierachical clustering to identify the various core structures. Then 3D protein-ligand interaction fingerprints were used as input. The authors also identified important features, e.g., specific vdW interactions on key residues.
OnionNet: a Multiple-Layer Intermolecular-Contact-Based Convolutional Neural Network for Protein−Ligand Binding Affinity Prediction
Liangzhen Zheng, Jingrong Fan, and Yuguang Mu. (2019)
The authors from Nanyang Tech Univ developed OnionNet, a deep CNN model, to predict binding affinity of ligands to proteins given the structural information known. For featurization, the authors used element-pair-specific contacts between ligands and protein atoms, and grouped the contacts into different distance ranges, to encode the atom local environment. Three 2D-convolution layers were used without maxpooling. The model was benchmarked on PDBbind dataset and outperformed Pafnucy and RF-Score.
TF3P: Three-dimensional Force Fields Fingerprint Learned by Deep Capsular Network
Yanxing Wang, Jianxing Hu, Junyong Lai, Yibo Li, Hongwei Jin, Lihe Zhang, Liangren Zhang, Zhenming Liu. (2019)
The authors from Peking Univ developed a 3D fingerprint method by learning the latent space in capsular network. The capsNet contains encoder and decoder parts. The inputs are based on grid and use alkana carbon and proton to probe the vdW and eletrostatic potential. The fingerprints learned by TF3P are sensitive to 3D conformational change.
DeepAtom: A Framework for Protein-Ligand Binding Affinity Prediction
Yanjun Li, Mohammad A. Rezaei, Chenglong Li, Xiaolin Li, and Dapeng Wu. (2019)
The authors from Univ of Florida developed DeepAtom, a CNN-based framework to predict binding affinity. The input features of protein-ligand complex are atom types, e.g, H-bond donor/acceptor, positive/negative, hydrophobic etc, and volumn features. 3D CNN with maxpooling and shuffle groups are used in the model. The authors benchmarked on PDBbind dataset.
GraphAF: a Flow-Based Autoregressive Model for Molecular Graph Generation
Chence Shi, Minkai Xu, Zhaocheng Zhu, Weinan Zhang, Ming Zhang, Jian Tang. (2020)
This paper from CIFAR, Peking U and Shanghai Jiao Tong Univ developed a flow-based autoregressive model for generating molecular graph. The model defines an invertible transformation from a base distribution to a molecular graph, uses GCN to learn the graph representation, and sequentially generate nodes and edges for the molecules. The model is also able to fine tune molecular properties by taking these predictions into the reward function of reinforcement learning.
Active Learning Accelerates Ab Initio Molecular Dynamics on Pericyclic Reactive Energy Surfaces
Shi Jun Ang, Wujie Wang, Daniel Schwalbe-Koda, Simon Axelrod, and Rafael Gómez-Bombarelli. (2020)
The authors from MIT uses active learning to simualte a complicated reactive PES with multiple products and post-transition state bifurcation. The simulation predict a reaction mechanism that is in agreement with the experimentally-reported product distribution and suggest that post-transition state bifurcation plays a very minor role in the reaction. This overall approach is broadly applicable and opens the door to the study of dynamical effects in larger, previously-intractable reactive systems.